|
Asma
Resposta Tardia da Asma
LINFÓCITOS
Os linfócitos contribuem para a imunopatologia da asma, sobretudo através das células Th2, que sintetizam citocinas que participam do processo da elaboração da IgE, maturação e ativação de mastócitos e basófilos, assim como pela infiltração eosinofílica mediada via IL-5, determinando dano epitelial e hiper-responsividade brônquica. Alguns estudos também demonstraram citocinas dos linfócitos Th1 no soro e no lavado broncoalveolar (LBA) de pacientes com asma, especialmente durante as exacerbações da doença, embora a grande maioria dos estudos evidencie predomínio de citocinas Th2.1
O tráfego dos linfócitos nos pulmões ocorre continuamente através de dois sistemas funcionalmente distintos:  BALT (bronchus-associated lymphoid tissue) onde os antígenos inicialmente penetram no sistema e iniciam a resposta imune e o restante do parênquima pulmonar onde células diferenciadas T e B de memória, que se desenvolveram nos folículos secundários, transitam para interagir com os antígenos. As respostas humorais imunes induzidas pela BALT são principalmente a secreção de IgA.2 O aumento nos linfócitos T de memória após provocação antigênica pode ser consequência de uma proliferação local ou migração do MALT (mucosa-associated lymphoid tissue) e de linfonodos que drenam o parênquima regional pulmonar. BALT (bronchus-associated lymphoid tissue) onde os antígenos inicialmente penetram no sistema e iniciam a resposta imune e o restante do parênquima pulmonar onde células diferenciadas T e B de memória, que se desenvolveram nos folículos secundários, transitam para interagir com os antígenos. As respostas humorais imunes induzidas pela BALT são principalmente a secreção de IgA.2 O aumento nos linfócitos T de memória após provocação antigênica pode ser consequência de uma proliferação local ou migração do MALT (mucosa-associated lymphoid tissue) e de linfonodos que drenam o parênquima regional pulmonar.
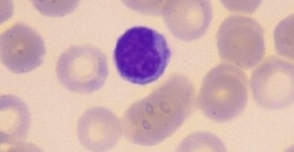
Os linfócitos apresentam uma participação muito importante na asma. Representam 20–40% das células brancas sanguíneas circulantes e 99% das células na linfa. São células redondas que pertencem ao grupo de mononucleares. Isso significa que o núcleo está inteiro. São células menores que os outros elementos do sangue, com um grande núcleo também redondo o qual é em geral ligeiramente excêntrico e com fino citoplasma (Figura 1). Ausência de nucléolos.3 Embora todos os linfócitos sejam morfologicamente semelhantes e tenham uma aparência bastante comum, eles são extremamente heterogêneos em linhagem, função e fenótipo, sendo capazes de respostas e atividades biológicas complexas
Podem ser classificados de variadas formas, dependendo de sua origem, de sua função ou dos marcadores de superfície (CD – Cluster of Differentiation) que portam. A classificação mais aceita e funcional é a Classificação por Linhagem e Função. É a divisão clássica entre imunidade adaptativa (específica) e inata (rápida) (Tabela 1). Linfócitos T (T de Timo) auxiliares ou Helper (CD4+); Linfócito B (Bone Marrow / medula óssea) e Células Natural Killer (NK).
Tabela 1 – Classificação dos Linfócitos
Tipo de Célula |
Marcador Principal |
Função Primária |
Alvo |
| Linfócito T (CD4+) |
CD3+, CD4+ |
Coordenação Imunológica |
Citocinas e ativação de outras células |
| Linfócito T (CD8+) |
CD3+, CD8+ |
Citotoxicidade |
Células infectadas e tumorais |
| Linfócito B |
CD 19+, CD20+ |
Produção de Anticorpos |
Patógenos extracelulares |
| Células NK |
CD 56+, CD16+ |
Lise Direta (Inata) |
Células "estranhas" sem MHC |
Diferentes linhagens ou diferentes estágios de maturação dos linfócitos podem ser caracterizados através da expressão de moléculas da membrana (proteínas de superfície), reconhecidas por anticorpos monoclonais específicos. Estas moléculas são chamadas de moléculas CD (cluster of differentiation), que definem um determinado tipo de célula ou estágio de diferenciação celular ou grupo de anticorpos, sendo contabilizadas atualmente mais de 371 tipos. Os linfócitos são classificados pelas moléculas que expressam em sua superfície e cada vez mais no caso das células T são subclassificados pelas citocinas que secretam. Por sua vez, essas moléculas de superfície e citocinas medeiam amplamente suas funções altamente especializadas.4 As chamadas células nulas constituem um pequeno grupo de linfócitos do sangue periférico que não expressam imunoglobulinas de membrana nem receptores de células T (TCR), correspondendo em grande parte, às células NK. Representam 10–15% dos linfócitos do sangue periférico e é um dos principais componentes da imunidade adaptativa, importantes na defesa contra infecções virais, respondendo rapidamente a patógenos sem ativação prévia. As células NK são altamente granulares devido ao armazenamento das enzimas líticas perforina e granzimas e destroem uma gama limitada de alvos ligados a IgG p. ex. células epiteliais infectadas com vírus, em uma reação de citotoxicidade dependente de anticorpos.4
Respostas imunológicas adaptativas requerem linfócitos. Respostas imunes inatas se beneficiam das respostas imediatas e não específicas dos neutrófilos, macrófagos e de populações de linfócitos que respondem precocemente aos antígenos estranhos.5
Dentre os leucócitos, somente os linfócitos T são capazes de iniciar respostas imunes após o reconhecimento de antígenos externos através de receptores antígeno específicos, exercendo também funções como células apresentadoras de antígenos (APCs). Na atualidade já existe considerável conhecimento para a aceitação da hipótese de que a asma represente uma forma especializada de imunidade celular mediada, na qual citocinas e possivelmente outros mediadores secretados e ativados pelos linfócitos T promovam acumulação específica e ativação de eosinófilos na mucosa brônquica.
Nos brônquios, tanto de pacientes com asma atópica como na não atópica, têm sido encontrados linfócitos T auxiliares ativados, que expressam o receptor IL-2 (CD25+) em sua superfície.6-8 O número de linfócitos ativados correlaciona-se com a gravidade da doença, com o número de eosinófilos ativados, com o valor do Pico de Fluxo Expiratório (PFE)9 e com a concentração de metacolina necessária para produzir queda de 20% (PC20) no VEF1.
Quanto ao estado de ativação os linfócitos são classificados como:
● |
Naïve (vírgens): linfócitos que estão maduros, mas ainda não encontraram seu antígeno específico; |
|
|
● |
Efetores: células que executam imediatamente a resposta imune após ser ativado pelo encontro com seu antígeno específico; |
|
|
● |
Memória: é uma célula de longa duração gerada durante a resposta imune primária, que sobrevive para garantir proteção a longo prazo. |
|
|
Assim, os linfócitos ativados são células muito importantes na resposta imune, coordenando e amplificando a atividade efetora antígeno específica e não específica de células inflamatórias, como os linfócitos B e os eosinófilos. Em síntese, os linfócitos T se dividem em duas categorias distintas de acordo com os marcadores de superfície e atividade. Os que expressam o antígeno CD4+ participam principalmente da imunidade humoral e são chamados de T auxiliares, incitando a produção de IgE e IgG4, enquanto que os que expressam o antígeno CD8+ são chamados de T supressores, participam da resposta imune celular e síntese de IgG2a e interagem com o complexo principal de histocompatibilidade (MHC) classe I.10
No LBA de adultos saudáveis, não fumantes, sem doença pulmonar, as células que predominam são os macrófagos > 85%, linfócitos (CD4+/CD8+) 10–15%, neutrófilos ≤ 3%, eosinófilos ≤ 1% e ≤ 5% de células epiteliais/colunares.11 Aproximadamente 12,8 X 103 linfócitos são recolhidos quando de LBA em indivíduos normais.12 São geralmente células T de memória CD54R0 que coexpressam o receptor de célula T a/ß (TCR). Por outro lado, apenas um pequeno número de células normais pulmonares (5%) se 'coram' com o anticorpo monoclonal TCRδ1 que reconhece o epítopo comum da cadeia d expresso por todas as células TCR γ/δ. Na asma ocorrem contagens diferenciais de células no LBA com mais de 1% de eosinófilos e mais de 0,5% de mastócitos.11
Citocinas
Os linfócitos T apresentam papel essencial na iniciação e regulação das respostas inflamatórias, pois ajudam a ativar as respostas de outras células, através da secreção de uma variedade de mediadores locais, coletivamente denominados de citocinas. O termo citocina, proposto em 1974 por Cohen et al.13 inclui linfocinas e monocinas, palavras não mais usadas na atualidade. As citocinas representam uma linguagem universal no diálogo entre as diferentes células do organismo. As citocinas são proteínas de baixo peso molecular e funcionam como moléculas mensageiras do sistema imunológico. Em geral atuam localmente em tecidos contíguos, de forma gradiente-dependente. Qualquer célula, cuja atividade é modificada seguindo a mensagem de uma citocina, possui um receptor específico em sua superfície. Podem apresentar atividade sistêmica como agentes endócrinos. São secretadas por uma variedade de células e atuam sobre outras células, ligando-se a receptores de citocinas. Esta ligação de alta afinidade entre a citocina e seu receptor permite que pequenas quantidades da substância apresentem ação potente. Quando a citocina se liga ao receptor, ele emite sinais intracelulares (sinais de transdução) que conduzem a específicas modificações na expressão de genes. Muitas das citocinas inicialmente eram designadas de interleucinas (IL) pois atuam na comunicação entre os leucócitos.14
As citocinas produzidas pelos linfócitos T têm participação importante na resposta inflamatória crônica da asma. Atuam em receptores específicos de células-alvo, transmitindo sinais que determinam ativação, proliferação, quimiotaxia, imunomodulação, ativação de outras citocinas ou mediadores, crescimento e diferenciação celular e apoptose. Apresentam também participação na regulação da expressão de moléculas de adesão, tanto nas células endoteliais da circulação brônquica como nas células epiteliais.15,16
Os linfócitos possuem vida longa e memória imunológica e também podem ser ativados por antígenos específicos. Sua importância na doença é sugerida pelo aumento de CD4+ ativados encontrados no LBA, em biópsias brônquicas e no sangue periférico.
Os linfócitos produzem muitas interleucinas, entretanto, cada subtipo apresenta um "perfil" bem característico (Tabela 2).
Neste capítulo se dará ênfase sobretudo aos linfócitos T (CD4+ e CD8+) e linfócitos B que são as principais fontes linfocitárias.
Tabela 2 – Citocinas Produzidas de Acordo com o Perfil dos Linfócitos
PRODUÇÃO DE CITOCINAS POR LINFÓCITOS |
Eixo/Subtipo |
Citocinas Produzidas |
Apontamento Funcional |
|
| Th1 (CD4+) |
IFN-γ, IL-2, TNF-α |
Imunidade celular, ativação macrofágica, inflamação clássica |
Papel modulador, antagoniza Th2, pode coexistir com Th17 |
| Th2 (CD4+) |
IL-4, IL-5, IL-9, IL-13, IL-10 |
Eosinofilia, IgE, muco, hiper-responsividade |
Asma T2-high, AVT eosinofílica |
| Th17 (CD4+) |
IL-17A, IL-17F, IL-21, IL-22, IL-26 |
Neutrofilia, disfunção epitelial, córtico-resistência |
Asma não T2, AVT refratária |
| Tfh (CD4+) |
IL-21, IL-4, IL-10 |
Ajuda a linfócito B, IgE |
Asma atópica |
| Treg (CD4+) |
IL-10, IL-35 |
Supressão imunológica |
Deficiência ⇒ asma grave |
| T CD8+ |
IL-2, TNF-β, IFN-γ, IL-17* |
Citotoxicidade, inflamação mista |
Exacerbações, asma mista (Tc17) |
| Linfócito B |
IL-6, IL-10, IL-35 |
Amplificação Th2 ou regulação |
Persistência / regulação |
| Tγδ |
IL-17, IL-22, IL-10 |
Resposta rápida tipo inata |
Exacerbações dano epitelial |
A IL-4 apresenta múltipla e relevante participação na fisiopatologia da asma, mencionando-se:
➢ inibe a apoptose de eosinófilos e promove a inflamação eosinofílica promovendo a quimiotaxia de eosinófilos e ativação por meio do aumento da expressão de eotaxina;17
➢ é um cofator com a Stem Cell Factor (SCF) que promove a proliferação e diferenciação de mastócitos;18
➢ em estudos humanos, ela induz a síntese de IgE pelos linfócitos B. A capacidade dos clones de células T em apoiar a produção de IgE é diretamente proporcional a sua produção de IL-4;19
➢ no decurso da inflamação alérgica tem a capacidade de conduzir a diferenciação de linfócitos T helper tipo 0 (Th0) naïve em linfócitos Th2;20,21
➢ upregulation da FcεRII e a atuação na expressão antigênica do complexo maior de histocompatibilidade II (MHC classe II) sobre as células apresentadoras de antígenos (APCs);
➢ upregulation da expressão da vascular cell adhesion molecule (VCAM-1) nas células endoteliais;18
➢ indivíduos atópicos têm um maior número de células T produtoras de IL-4 em comparação com indivíduos normais;22
➢ a IL-4 contribui para a obstrução das vias aéreas na asma por meio da indução da expressão do gene da mucina e da hipersecreção de muco;23
➢ a IL-4 aumenta a expressão de eotaxina e outras citocinas inflamatórias de fibroblastos que podem contribuir para a inflamação e remodelamento pulmonar na asma crônica;24
Estudos efetuados através de biópsia brônquica evidenciaram ao nível do epitélio e subepitélio, o aumento na expressão da IL-4, tanto sob a forma proteica como sob a forma mRNA, em pacientes com asma atópica e não atópica, o que não foi demonstrado em controles não asmáticos.20,21,25 A IL-4 parece ser um pré-requisito para que os linfócitos T iniciem a produção da IL-5.26
IL-5 A IL-5 é o principal fator de crescimento, diferenciação, ativação, adesão, e sobrevida dos eosinófilos, bloqueando sua apoptose.27,28 Em condições alérgicas atua como uma eosinofilopoetina. A administração exógena de IL-5 ocasiona eosinofilia em uma variedade de modelos in vivo. Por outro lado, a expressão da IL-5mRNA correlaciona-se com os índices clínicos de gravidade da asma.29,30 Outros trabalhos, através de biópsias brônquicas, comprovaram que a expressão do receptor IL-5 é restrita praticamente aos eosinófilos (> 90%), enquanto que a expressão IL-5Ra na membrana celular correlaciona-se inversamente com o VEF1 basal, ao passo que a expressão do IL-5Ra solúvel (sIL-5Ra), com ação antagônica a IL-5, correlaciona-se positivamente com o VEF1.31 Em ratos transgênicos a expressão aumentada de IL-5 no epitélio respiratório resulta em maior hiper-responsividade brônquica. A IL-5 promove o acúmulo de eosinófilos nas doenças alérgicas inflamatórias através da upregulation de respostas de quimiocinas e integrinas adß2 nos eosinófilos, promovendo desta forma sua aderência a VCAM-1 expressa nas células endoteliais e subsequente migração transendotelial.32 Também estimula linfócitos B. É uma citocina sintetizada principalmente por linfócitos Th2 e mastócitos. 15,33
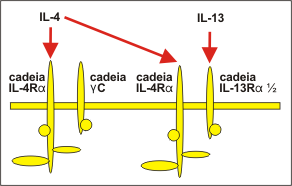 IL-13 A IL-13 é uma citocina pleiotrópica que tem papel crítico e central para a patogênese da inflamação Th2 da asma.34,35 Um aumento na expressão de IL-13 tem sido descrito tanto na asma atópica como na não atópica, após provocação antigênica. A IL-13 tem aproximadamente 30% de homologia com a IL-4 e compartilha muitas de suas atividades biológicas em células fagocíticas mononucleares, células endoteliais, células epiteliais e células B. Ambas têm atividades biológicas muito parecidas, em decorrência da estrutura de seus receptores. O receptor da IL-13 consiste nas cadeias IL-13Ra 1 ou a 2 que se ligam à IL-13, apresentando ainda, uma cadeia IL-4Ra. A IL-4 pode, portanto, ligar-se a ambos os receptores (IL-4 e IL-13) através da cadeia IL-4Ra, enquanto que a IL-13 se liga somente ao seu próprio receptor (Figura 2).18 A IL-13 é uma citocina secretada por células Th2, outras células CD4+, células NK, mastócitos, basófilos, eosinófilos e ILC2. IL-13 A IL-13 é uma citocina pleiotrópica que tem papel crítico e central para a patogênese da inflamação Th2 da asma.34,35 Um aumento na expressão de IL-13 tem sido descrito tanto na asma atópica como na não atópica, após provocação antigênica. A IL-13 tem aproximadamente 30% de homologia com a IL-4 e compartilha muitas de suas atividades biológicas em células fagocíticas mononucleares, células endoteliais, células epiteliais e células B. Ambas têm atividades biológicas muito parecidas, em decorrência da estrutura de seus receptores. O receptor da IL-13 consiste nas cadeias IL-13Ra 1 ou a 2 que se ligam à IL-13, apresentando ainda, uma cadeia IL-4Ra. A IL-4 pode, portanto, ligar-se a ambos os receptores (IL-4 e IL-13) através da cadeia IL-4Ra, enquanto que a IL-13 se liga somente ao seu próprio receptor (Figura 2).18 A IL-13 é uma citocina secretada por células Th2, outras células CD4+, células NK, mastócitos, basófilos, eosinófilos e ILC2.
A IL-13 desempenha um papel central na resposta imune, promovendo a diferenciação, proliferação e troca de isotipo das células B, favorecendo a produção de IgE. Além disso, estimula o desenvolvimento de células dendríticas e contribui para o acúmulo de eosinófilos por aumento da expressão de VCAM-1 nas células endoteliais.36
Alguns dos efeitos mais proeminentes da IL-13 incluem aumento na diferenciação das células caliciformes secretoras de muco (hiperplasia de células caliciformes e hipersecreção de muco), produção de proteínas de matriz extracelular e diferenciação de miofibroblastos, elevação da hiper-responsividade brônquica, aumento da contratilidade de células musculares lisas das vias aéreas em resposta a agonistas colinérgicos e desvio na produção de anticorpos de células B de IgM para IgE.37 A IL-13 também estimula a secreção de periostina, uma proteína matricelular, que tem um papel na ativação dos fibroblastos e no aumento da elasticidade do gel de colágeno.38 Ao ativar a sintase do óxido nítrico epitelial por meio de seu efeito sobre a signal transducer and activator of transcription 6 (STAT 6), sua atividade biológica também pode ser refletida por níveis elevados de óxido nítrico exalado (FeNO).39
As quimiocinas são indispensáveis para o recrutamento de células para o local da inflamação. O termo quimiocina foi proposto em 1992 para definir esta família de proteínas de baixo peso molecular com 6-10 kDa com similaridade de 20–55% na sequência de aminoácidos, sendo relativamente celular específica.40 Até o momento foram identificadas mais de 50 quimiocinas e são 20 os receptores acoplados à proteína G. Estas moléculas foram divididas em quatro grupos estruturais baseados na organização dos resíduos de cisteína próximos ao N-terminal: CXC, CC, C e CX3C. As duas maiores famílias são a CXC e a CC.
Os nomes aplicados historicamente aos ligantes frequentemente idênticos das quimiocinas foram condensados após um consenso, em duas grandes subfamílias: de CCL1 a CCL28 e CXCL1 a CXCL16, bem como em duas pequenas subfamílias: XCL1 e XCL2 e CX3CL1, em função da identificação de um resíduo cisteína. Os receptores de quimiocinas estão distribuídos entre duas grandes subfamílias, CCR1 a CCR10 e CXCR1 a CXCR6, assim como duas pequenas subfamílias, XCR1 e CX3CR1.41
A inflamação alérgica é regulada pelas células T, do subtipo Th2. O tráfego e recrutamento de células Th2 para os sítios de inflamação depende da expressão de receptores para quimiocinas CC e CD4. As células T que se diferenciam in vitro no fenótipo Th2 expressam os receptores de quimiocinas CCR3, CCR4 e CCR842-45 e interagem com seus ligantes: CCL11 (eotaxina-1), CCL22 (monocyte-derived chemokine (MDC)), CCL17 (Thymus-and activation-regulated chemokine (TARC)), e CCL1 (I-309/TCA-3). O CCR4 é fortemente expresso em células Th2 ativadas in vitro. Além disso, um estudo atesta a expressão CCR4 em células T que infiltram as vias aéreas após teste de provocação alérgica em pacientes com asma. Outra publicação demonstra um aumento na CCR4 nos linfócitos do sangue periférico de pacientes com dermatite atópica, quando comparado a normais.46
Desta forma, antagonistas CCR3 poderiam modular o recrutamento de células Th2 e consequentemente regular a inflamação alérgica na asma. Existem evidências de que a quimiocina MCP-1, sinalizando via receptor CCR2, apresente importante participação na regulação da diferenciação Th2 /Th1.
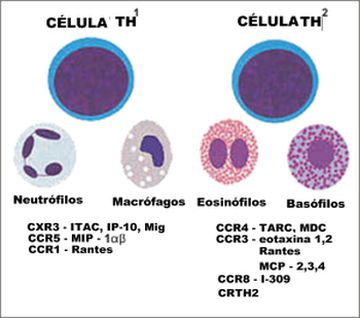
Outro papel das quimiocinas consiste na coordenação dos vários tipos de células que participam das respostas inflamatórias, o que pode ser chamado como "unidades funcionais imunes". As células Th1 colocalizam macrófagos e neutrófilos nos sítios periféricos inflamatórios, enquanto que os eosinófilos e basófilos são encontrados juntos a células Th2. Pelo perfil de produção de citocinas e quimiocinas, as células Th1 e Th2 influenciam a ativação e migração de outras células inflamatórias, bem como de células teciduais residentes (Figura 3). A participação das quimiocinas Th2 é importante não só pelo simples recrutamento de uma combinação de células inflamatórias no microambiente, como na determinação das características morfológicas do infiltrado inflamatório.47
Células T Reguladoras (Tregs)
 A tolerância imunológica é a capacidade do sistema imunológico de não reagir contra certos antígenos. Esse mecanismo é essencial para impedir que o corpo ataque seus próprios tecidos (os chamados autoantígenos), evitando o desenvolvimento de doenças autoimunes. Em outras palavras, é o processo pelo qual o sistema imune aprende a diferenciar o que pertence ao organismo ("próprio") daquilo que é estranho ou potencialmente perigoso ("não próprio"). A tolerância imunológica é a capacidade do sistema imunológico de não reagir contra certos antígenos. Esse mecanismo é essencial para impedir que o corpo ataque seus próprios tecidos (os chamados autoantígenos), evitando o desenvolvimento de doenças autoimunes. Em outras palavras, é o processo pelo qual o sistema imune aprende a diferenciar o que pertence ao organismo ("próprio") daquilo que é estranho ou potencialmente perigoso ("não próprio").
Os Tregs são guardiões essenciais da tolerância na asma através de múltiplos mecanismos supressores. Em indivíduos saudáveis, a exposição a antígenos ambientais inofensivos não provoca uma resposta imune efetora prejudicial aos tecidos. Por padrão, esses antígenos são ignorados ou especificamente tolerados pelo sistema imunológico. De um ponto de vista mecanicista, sinais de perigo são necessários para a regulação positiva de moléculas coestimulatórias por APCs.48 A tolerância imunológica é um estado que previne reações contra antígenos inofensivos, prevenindo alergias e inflamação inadequada, minimizando danos.
Os linfócitos T reguladores (Tregs) se constituem em uma população especializada de células T CD4+ que promovem tolerância periférica, mantêm a homeostase e limitam a inflamação na asma, especialmente na forma alérgica (asma atópica) – são linfócitos que regulam outros linfócitos.49 Assim, as Tregs desempenham um papel importante na supressão do sistema imunológico e na geração de tolerância e na asma a sua função principal seria a de suprimir a inflamação alérgica e manter o equilíbrio entre imunidade e tolerância.50-54
Os Tregs mantêm a homeostase e limitam a inflamação crônica das doenças alérgicas assim como na asma a sua função principal seria a de
suprimir a inflamação alérgica e manter o equilíbrio entre imunidade e tolerância.49-53
A asma não resulta apenas de um sistema imunológico "hiperativo", mas também de falhas no "sistema de freios" representado pelas células Tregs. Em indivíduos asmáticos, há uma deficiência quantitativa ou funcional dessas células, o que leva a uma insuficiente supressão dos linfócitos Th2. Esses linfócitos produzem citocinas essenciais para a inflamação do tipo 2, desempenhando papel central na fisiopatologia da asma persistente e não controlada. Já em pessoas saudáveis, os Tregs mantêm os linfócitos Th2 sob controle, evitando respostas exageradas a antígenos inofensivos, como poeira ou pólen.
Os Tregs são um subconjunto de células T CD4+ e podem ser divididos, com base em sua origem, em Tregs naturais (nTregs) e Tregs induzidos (iTregs).55 Os nTregs são derivados de timócitos e também são conhecidos como Tregs derivados do timo (tTregs) e expressam o fator de transcrição forkhead box P3 (Foxp3), responsáveis pela autotolerância. Os Tregs induzidos perifericamente (pTregs) provêm de células convencionais (naïves) pela exposição ao antígeno (próprio ou externo) em um contexto de tolerância, ou na presença de citocinas imunossupressoras.56-58
Além dos Tregs naturais (origem no timo), os Tregs também são classificados com base em marcadores de superfície e perfil funcional. No pulmão, a tolerância imunológica é controlada principalmente por três subconjuntos de células Tregs:55 iTregs CD4+CD25+Foxp3+, que expressam o fator de transcrição específico Foxp3; células Th3 CD4+CD25lowFoxp3+, que secretam principalmente TGF-β e células T reguladoras tipo 1 (Tr1) CD4+CD25lowFoxp3−, que secretam altos níveis de IL-10, que desempenham um papel essencial no estabelecimento de uma resposta imune saudável e equilibrada a alérgenos.
Funções das Tregs na Asma:
As Tregs utilizam um arsenal variado para "desligar" a inflamação pulmonar:
1) Regulam a inflamação das vias aéreas através da liberação de diversas citocinas, incluindo:
● IL-10 (produzida por uma subdivisão pTreg FOXP3+, as células Tr1), que inibe a ativação de células dendríticas, impedindo que elas apresentem antígenos para novos linfócitos. Inibe a produção de citocinas Th2 (IL-4. IL-5 IL-13) assim como inibe ativação de eosinófilos e mastócitos.
Em síntese, por meio da liberação de IL-10, as Tregs modulam negativamente a resposta imune: inibem a síntese de citocinas pró-inflamatórias, reduzem a expressão de citocinas nas células T efetoras e limitam a apresentação de antígenos e a coestimulação pelas APCs.59
● TGF-β (produzidas predominantemente por células T helper tipo 3), que suprime a proliferação de céluas T efetoras do sistema inato e promove a manutenção da integridade epitelial.60
● IL-35 (em certos contextos – iTr-35) efeito adicional supressor em células T e B.
2) Expressão de moléculas reguladoras
➢ São moléculas reguladoras adicionais que atuam em conjunto e contribuem para modulação do ambiente inflamatório em doenças como a asma ampliando a função supressora dos Tregs.
● As células Tregs também podem modular a inflamação por meio da supressão mediada pelo contato celular Cytotoxic T-Lymphocyte Antigen 4
[CTLA-4].61
Liga-se com alta afinidade aos receptores CD80/CD86 em células apresentadoras de antígeno (APCs, principalmente células dendríticas), inibindo coestimulação. Assim a Treg impede que a célula dendrítica dê o "segundo sinal" necessário para ativar um linfócito T contra um alérgeno (pólen, ácaro).
● LAG-3 (Lymphocyte Activation Gene-3) – expressa por Tregs ativados, células T e algumas células NK. Na asma pode ser induzida em Tregs pulmonares cronicamente expostos a antígenos, contribuindo para conter a hiper-responsividade das vias aéreas.62 A LAG-3 se liga diretamente ao MHC-II, impedindo que a célula apresentadora de antígenos consiga acoplamento, silenciando a comunicação sináptica imunológica.
● Galectina-1 – promove apoptose de células Th1 e Th17. Na asma tem efeito protetor, suprimindo a inflamação alérgica, podendo favorecer a tolerância à exposição crônica a alérgenos.63
● PD-1/PD-L1 – esta via promove a exaustão dos linfócitos efetores. No contexto da asma, seu adequado funcionamento contribui para reduzir a atividade agressiva dos linfócitos T no parênquima pulmonar.
3) Consumo de IL-2
● A IL-2 é uma citocina fundamental para a proliferação de células T, sem a qual a célula T efetora (Th2 no caso da asma) não consegue se dividir, sobreviver muito tempo. As Tregs expressam níveis elevados de CD25 (a cadeia alfa do receptor de IL-2 de alta afinidade), competindo com células T efetoras por IL-2, agindo como uma "esponja". Elas captam a IL-2 do ambiente com muito mais efetividade. A IL-2 é uma citocina essencial para a homeostase e função dos Tregs. Os Tregs expressam constitutivamente altos níveis de CD25, e a IL-2 é essencial para preservar a tolerância, influenciando a homeostase e a ativação dos Tregs.64-66
As células Tregs modulam as respostas imunes alérgicas do tipo Th2 e exercem papel central no controle das doenças como a asma, atuando através de diversas vias regulatórias (Figura 4). As Tregs funcionam como um freio imunológico essencial, atuando em múltiplos pontos da resposta alérgica para restaurar o equilíbrio e prevenir uma reação exagerada contra alérgenos inofensivos. A sua disfunção ou baixa frequência está associada ao desenvolvimento e gravidade de doenças alérgicas, enquanto estratégias que expandem ou aumentam a sua função (como a imunoterapia específica) são a base para tratamentos capazes de modular ativamente as vias protetoras responsivas a alérgenos do sistema imunológico e alterar o curso natural da doença.
Células Linfoides Inatas (ILCs)
As células linfoides inatas (ILCs) são uma família crescente de células imunológicas que refletem os fenótipos e funções das células T. Muitos estudos sustentam que as células linfoides inatas 2 (ILC2) estão envolvidas na patogênese da asma, interagindo com células estruturais e com o sistema imunológico inato. Por décadas, a asma tem sido considerada como uma doença imunológica mediada por células Th2 e de imunidade adaptativa.67 Entretanto, outros fenótipos não alérgicos da asma associados à exposição a fatores ambientais, como poluição do ar, incluindo ozônio, partículas de diesel e fumaça de cigarro, exercício, infecção viral, estresse e obesidade estão frequentemente relacionados a neutrófilos nas vias aéreas e à imunidade inata independente de células Th2.68-72
Em 2008–2009, cerca de 12 grupos independentes relataram a identificação em mamíferos de novos tipos de linfócitos não T e não B. Essas células foram inicialmente chamadas de "nunócitos"73 e posteriormente de células linfoides inatas (ILCs). As ILCs são em grande parte células residentes nos tecidos e estão profundamente integradas na estrutura dos tecidos.74 Não expressam receptores de antígenos adquiridos mas respondem rapidamente a sinais de citocinas e outras moléculas presentes no microambiente tecidual. Elas têm participação na homeostase do tecido, na organogênese do tecido linfoide, na resistência à infecção, no controle da composição da microbiota comensal e na patologia das superfícies mucosas. Secretam uma variedade de citocinas, semelhantes aos linfócitos T como IFN-γ, IL-5, IL-13, IL-17 e IL-22.75
Observamos três grupos de ILCs, todos originários de células progenitoras linfoides comuns (Figura 5). ILC1 que produz IFN-γ e TNF-α conferindo-lhe propriedades de imunidade antiviral e antitumoral. As ILC2 produzem IL-4, IL-5, IL-9, IL-13 e anfiregulina, encontrando-se associadas a respostas imunológicas relacionadas a helmintos e na regulação de reações alérgicas. Por fim, as ILC3 estão correlacionadas a doenças autoimunes e secretam IL-17, IL-22 e GM-CSF.76
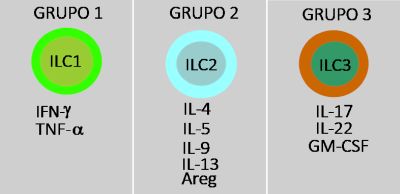
Figura 5 – As células linfoides inatas são uma família de células do sistema imunológico classificadas atualmente em três grupos principais de acordo com as citocinas que produzem e em função das respostas que desencadeiam: ILC1 – defesa contra patógenos intracelulares como vírus e bactérias; ILC2 – atuam na resposta orgânica contra parasitas como os helmintos e são determinantes na patogênese das doenças alérgicas como a asma; ILC3 – participam na defesa contra patógenos extracelulares tais como fungos e bactérias especialmente nas barreiras mucosas como os intestinos.
As ILC2s implicadas em doenças alérgicas e na asma são particularmente definidas pela expressão dos fatores de transcrição GATA3, RORα, BCL11B e GFI1, em geral, proliferam em resposta à TSLP, IL-25 e IL-33 e secretam citocinas do tipo 2 (principalmente IL-5, IL-9, IL-13, mas não IL-4)77 e anfiregulina (AREG).76 Tanto o GATA3 quanto o BCL11B desempenham papéis críticos no desenvolvimento de células T e ILC2s. ILC2s contêm quantidades maiores do fator de transcrição GATA3 do que os outros subconjuntos ILCs e ausência de GATA3 inibe o desenvolvimento e a função dessas células.78-80
A ILC2 é a população predominante de ILCs no pulmão, em contraste com outras superfícies mucosas.81 As ILC2s  desempenham importante papel no processo da asma alérgica, pois são ativadas a jusante das células epiteliais das vias aéreas. As ILC2s são recrutadas para locais de inflamação pulmonar por células epiteliais das vias aéreas, macrófagos alveolares e células dendríticas que liberam as quimiocinas CCL17 e CCL22, ambas atuando através do receptor CCR4 nas ILC2s. Diante de estímulos externos como alérgenos, vírus, bactérias, fumaça de cigarro e poluentes do ar, as células epiteliais respondem gerando várias citocinas, e particularmente um trio de citocinas epiteliais conhecidas como alarminas – IL-25, IL-33 e TSLP. Essas citocinas estimulam respostas inflamatórias por meio de inúmeras vias na asma que demonstraram iniciar respostas de células ILC2 que podem produzir enormes níveis de citocinas do tipo 2 que modulam funções das células efetoras, como IL-5 e IL-13 assim como IL-9 e anfiregulina, tanto em murinos como em humanos.82 desempenham importante papel no processo da asma alérgica, pois são ativadas a jusante das células epiteliais das vias aéreas. As ILC2s são recrutadas para locais de inflamação pulmonar por células epiteliais das vias aéreas, macrófagos alveolares e células dendríticas que liberam as quimiocinas CCL17 e CCL22, ambas atuando através do receptor CCR4 nas ILC2s. Diante de estímulos externos como alérgenos, vírus, bactérias, fumaça de cigarro e poluentes do ar, as células epiteliais respondem gerando várias citocinas, e particularmente um trio de citocinas epiteliais conhecidas como alarminas – IL-25, IL-33 e TSLP. Essas citocinas estimulam respostas inflamatórias por meio de inúmeras vias na asma que demonstraram iniciar respostas de células ILC2 que podem produzir enormes níveis de citocinas do tipo 2 que modulam funções das células efetoras, como IL-5 e IL-13 assim como IL-9 e anfiregulina, tanto em murinos como em humanos.82
Essas citocinas inatas, geradoras de respostas do tipo 2, podem ser liberadas em resposta à inalação de alérgenos, desencadeando alterações patológicas nos pulmões que são características da inflamação alérgica, com suas consequências fisiopatológicas. Além disso, relatos indicam que outras substâncias solúveis podem ativar as ILC2s, incluindo os leucotrienos.83 A IL-5 produzida pelas ILC2s exerce dois efeitos principais na asma: no sangue, envia sinais à medula óssea que estimulam a eosinopoiese;84,85 no pulmão e nas vias aéreas,. atua em conjunto com moléculas quimioatraentes, como o leucotrieno B4, favorecendo a migração de eosinófilos para o local da inflamação.84,86 Por outro lado, a IL-13 é importante para a remodelação das vias aéreas: induz à diferenciação das células Clara em células caliciformes, o que conduz à hiperplasia dessas células e ao aumento da secreção de muco, favorecendo a obstrução característica da asma.87-89
Uma de suas características marcantes é a produção significativa de PGD2, induzindo a ativação autócrina das ILC2s por meio do receptor CRTh2.90,91 As principais fontes de PGD2 no pulmão são mastócitos e ILC2s, que, portanto, podem se autoestimular de forma parácrina ou autócrina.92 A ativação das ILC2s é caracterizada pela sua expansão e pela produção de citocinas do tipo 2, incluindo a IL-4 em humanos.76 Essa expansão das ILC2s pode ser alcançada tanto pelo recrutamento de ILC2s para o local da provocação quanto pela proliferação in loco das células residentes.93 As ILC2s fazem muitas coisas em comum com as células Th2, mas são únicas em sua alta secreção de IL-9, com potencial em promover a metaplasia de células caliciformes e promover o crescimento e a sobrevivência dos mastócitos.94
Foi demonstrado que a AREG produzida por ILC2s controla a proliferação e diferenciação de células epiteliais necessárias para o reparo após a infecção por vírus influenza.95,96 A anfiregulina produzida por várias células, incluindo as ILC2s, pode desempenhar um papel nos processos de reparação e remodelação observados, provavelmente, também na asma.
As ILC2s também estão associadas a várias patologias, dentre elas, a fibrose pulmonar,97 a rinossinusite crônica,98 a dermatite atópica,99,100 além de alergias100,101 e exacerbações de asma induzidas em pacientes por rinovírus.102,103
|
|
Home
 Design by Walter Serralheiro Design by Walter Serralheiro
|
|
Referências
01.Corrigan CJ, Kay AB. – T Lymphocytes in AsThma PaThogenesis. In: P.J. Barnes, M.M. Grunstein, A.R. Leff. and A.J. Woolcock. AsThma. Philadelphia: Lippincott-Raven Publishers; 1997:433-451.
02.Bienenstock J, McDermott MR. Bronchus- and nasal-associated lymphoid tissues. Immunol Rev 2005; 206:22-31.
03.Oliveira. HP. – Hematologia Clínica. 1ª edição. Rio de Janeiro: ATheneu; 1978.
04.Anderson GP. – Lymphocytes. In: P.J. Barnes, I.W.Rodger, N.C.Thomson. Asthma. 3rd Ed. San Diego: Academic Press; 1998:159-186.
05.Cohn L,
Hawrylowicz C,
Anuradha R. – Biology of Lymphocytes. In:
N.F. Adkinson Jr, B.S. Bochner, W. Burks. Middleton's Allergy: Principles and Practice. 8Th ed.
Philadelphia: Saunders; 2014:203-214.
06.Azzawi M, Bradley B, Jeffery PK, et al. Identification of actived T lymphocites and eosinophils in bronchial biopses in stable atopic asThma. Am Rev Respir Dis 1990; 142:1407-13.
07.Bradley BL, Azzawi M, Jacobson M, et al. Eosinophils, T-lymphocites, mast cells, neutrophils, and macrophages in bronchial biopsy specimens from atopic subjects wiTh asThma: Comparison wiThbiopsy specimens from atopic subjects wiThout asThma and normal control subjects and relationship to bronchial hyperresponsiveness. J Allergy Clin Immunol 1991; 88:661-74.
08.Hamid Q, Barkans J, Robinson DS et al. Co-expression of CD25 and CD3 in atopic allergy and asThma. Immunology 1992; 75:659-63.
09.Corrigan CJ, Hartnell A, Kay AB. T lymphocyte activation in acute severe asThma. Lancet 1988; 1:1129-32.
10.Bostoff J, Scadding GK, Male DK, Roitt IM. – Immunologie Clinique.4eme ed. Paris: De Boeck & Larcier; 1997.
11.Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, Rottoli P, Saltini C, Selman M, Strange C, Wood B; American Thoracic Society Committee on BAL in Interstitial Lung Disease. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med 2012; 185:1004-14.
12.Foresi A, Bertorelli G, Pesci A, Chetta A, Olivier D. Inflammatory Markers in Bronchoalveolar Lavage and in Bronchial Biopsy in AsThma during Remission. Chest 1990; 98;528-535.
13.Cohen S, Bigazzi PE, Yoshida T. Similarities of T cell in function cell-mediated immunity and antibody production. Cell Immunol 1974; 12:150-59.
14.Cavaillon JM. – Les Cytokines. 2ª ed. Paris: Masson; 1996.
15. Krouwels FH, Hol BE, Bruinier B, Lutter R, Jansen HM, Out TA. Cytokine production by T-cell clones from bronchoalveolar lavage fluid of patients with asthma and healthy subjects. Eur Respir J Suppl 1996; 22:95s-103s.
16.Lambrecht BN, Hammad H, Fahy JV. The Cytokines of Asthma. Immunity 2019; 50:975-991.
17.Nelms K, Keegam AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signalimg mechanisms and biologic functions. Annu Rev Immunol 1999; 17:701-38.
18.Borish L and Rosenwasser Lj. ? Cytokines in Allergic Inflammation. In: Busse WW, Bochner BS, Holgate ST, Simmons FE, Lemanske RF. Middleton's Allergy: Principles and Practice. 7Th ed. Philadelphia: Elsevier HealTh Sciences; 2008:165-179.
19.Del Prete G, Maggi E, Parronchi P, Chrétien I, Tiri A, Macchia D, Ricci M, Banchereau J, De Vries J, Romagnani S. IL-4 is an essential factor for The IgE synThesis induced in vitro by human T cell clones and Their supernatants. J Immunol 1988 15; 140:4193-8.
20.Humbert M, Durham SR, Ying S, Kimmitt P, Barkans J, Assoufi B, Pfister R, Menz G, Robinson DS, Kay AB, Corrigan CJ. IL-4 and IL-5 mRNA and protein in bronchial biopsies from patients wiTh atopic and nonatopic asThma: evidence against "intrinsic" asThma being a distinct immunopaThologic entity. Am J Respir Crit Care Med 1996; 154:1497-504.
21.Ying S, Humbert M, Barkans J, Corrigan CJ, Pfister R, Menz G, Larché M, Robinson DS, Durham SR, Kay AB. Expression of IL-4 and IL-5 mRNA and protein product by CD4+ and CD8+ T cells, eosinophils, and mast cells in bronchial biopsies obtained from atopic and nonatopic (intrinsic) asThmatics. J Immunol 1997; 1;158:3539-44.
22.Chan SC, Brown MA, Willcox TM, Li SH, Stevens SR, Tara D, Hanifin JM. Abnormal IL-4 gene expression by atopic dermatitis T lymphocytes is reflected in altered nuclear protein interactions wiTh IL-4 transcriptional regulatory element. J Invest Dermatol 1996; 106:1131-136.
23.Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, Nadel JA. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo . J Immunol. 1999; 162:6233-6237.
24.Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Jasmin C, Canonica GW, Azzarone B. IL-4 and IL-13 specifically increase adhesion molecule and inflammatory cytokine expression in human lung fibroblasts. Int Immunol 1998; 10:1421-1433.
25.Kotsimbos TC, Ghaffar O, Minshall EM, Humbert M, Durham SR, Pfister R, Menz G, Kay AB, Hamid QA. Expression of IL-4 receptor a -subunit is increased in bronchial biopsy specimens from atopic and nonatopic subjects. J Allergy Clin Immunol 1998; 102:859-66.
26.Steinke JW, Borish L. Th2 cytokines and asThma. Interleukin-4: its role in The paThogenesis of asThma, and targeting it for asThma treatment wiTh interleukin-4 receptor antagonists. Respir Res 2001; 2:66-70.
27.Chutterbuck EJ, Hirst EMA, Sanderson CJ. Human interleukin-5 (IL-5) regulates The production of eosinophils in human bone marrow cultures: comparison and interaction wiTh IL-1, IL-3, IL-6 and GM-CSF. Blood 1989; 73:1504-12.
28.Kroegel C, Virchow JC, Luttmann W, Walker C, Warner JA. Pulmonary immune cells in healTh and disease: The eosinophil leucocyte (part I). Eur Respir J 1994; 7:519-43.
29.Hamid Q, Azzawi M, Ying S et al. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asThma. J Clin Invest 1991; 87:1541-6.
30.Robinson DS, Ying S, Bentley AM et al. Relationships among numbers of bronchoalveolar lavage cells expressing messeger ribonucleic acid for cytokines, asThma symptoms, and metacholine responsiveness in atopic asThma. J Allergy Clin Immunol 1993; 92:397-403.
31.Yasruel Z, Humbert M, Kotsimbos TC et al. Membrane-bound and soluble a IL-5 receptor mRNA in The bronchial mucosa os atopic and nonatopic asThmatics. Am J Respir Crit Care Med 1997; 155:1413-18.
32.RoThenberg ME, Petersen J, Stevens RL, Silberstein DS, McKenzie DT, Austen KF, Owen WF Jr. IL-5-dependent conversion of normodense human eosinophils to The hypodense phenotype uses 3T3 fibroblasts for enhanced viability, accelerated hypodensity, and sustained antibody-dependent cytotoxicity. J Immunol 1989; 143(7):2311-6.
33.Okayama Y, Petit-Frére C, Kassel O, Semper A, Quint D, Tunon-de-Lara MJ, Bradding P, Holgate ST, Church MK. IgE-dependent expression of mRNA for IL-4 and IL-5 in human lung mast cells. J Immunol 1995; 155:1796-808.
34.Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, et al. Requirement for IL-13 independently of il-4 in experimental asThma. Science 1998; 282:2261-2263.
35.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergicasThma. Science 1998; 282:2258-2261.
36.Lordan JL, Hellewell PG. – Cytokines, Chemokines, and Adhesion Proteins. In: Holgate ST, Church MK, Lichtenstein LM. Allergy. London: Mosby; 2001:283-302.
37.Corren J. Role of Interleukin-13 in AsThma. Curr Allergy AsThma Rep 2013; 13(5):415-20.
38.Takayama G, Arima K, Kanaji T, et al. Periostin: a novel component of subepiThelial fibrosis of bronchial asThma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol 2006; 118 :98-104.
39.Chibana K, Trudeau JB, Mustovich AT, et al. IL-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synThase as compared wiTh effects on arginases in human primary bronchial epiThelial cells. Clin Exp Allergy 2008; 38:936-946.
40.Cavaillon JM. Interleukine-8 & Chémokines. In: J.-M.Cacaillon. Les Cytokines. Paris:Masson; 1996:217-231.
41.Murphy, K., Weaver, C. – Janeway's Immunobiology. 9ª ed. New York: Garland Science; 2017.
42.Gerber BO, Zanni MP, Ugoccioni M, Loetscher M, Mackay CR, Pichler WJ, Yawalkar N, Baggiolini M, Moser B. Functional expression of The eotaxin receptor CCR3 in T lymphocytes co-localizing wiTh eosinophils. Curr Biol 1997; 7:836-43.
43.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of The eotaxin receptor CCR3 by human T helper 2 cells. Science 1997; 277:2005-7.
44.Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998; 187:129-34.
45.D'Ambrosio D, Iellem A, Bonecchi R, Mazzeo D, Sozzani S, Mantovani A, Sinigaglia F. Selective up-regulation of chemokine receptors CCR4 and CCR8 upon activation of polarized human type 2 Th cells. J Immunol 1998; 161:5111-5.
46.Yamamoto J, Adachi Y, Onoue Y, Okabe Y, Itazawa T, Toyoda M, Seki T, Morohashi M, Matsushima K, Miyawaki T. Differential expression of The chemokine receptors by The Th1- and Th2-type effector populations wiThin circulating CD4+ T cells. J Leukoc Biol 2000; 68:568-74.
47.Sinigaglia F, Fabbri LM, D'Ambrosio D. Chemokine Receptors on Th1 and Th2 Cells. In : Hansel TT, Barnes PJ (eds). New Drugs for AsThma and COPD. Basel:Karger; 2001:284-287.
48.Matzinger P. The danger model: a renewed sense of self. Science 2002; 296(5566):301-5.
49.Zuany-Amorim C, Sawicka E, Manlius C, et al. Suppression of airway eosinophilia by killed Mycobacterium vaccae-induced allergen-specific regulatory T-cells. Nat Med 2002; 8:625-9.
50.Chiarella SE, Barnes PJ. Endogenous inhibitory mechanisms in asThma. J Allergy Clin Immunol Glob 2023; 2:100135. PMID: 37781649; PMCID: PMC10509980.
51.Khan MA. Regulatory T cells mediated immunomodulation during asThma: a Therapeutic standpoint. J Transl Med 2020;18:456.
52.Pellerin L, Jenks JA, Begin P, Bacchetta R, Nadeau KC. Regulatory T cells and Their roles in immune dysregulation and allergy. Immunol Res 2014; 58:358-68.
53.Zhang H, Kong H, Zeng X, Guo L, Sun X, He S. Subsets of regulatory T cells and Their roles in allergy. J Transl Med 2014; 12:125.
54.Thorburn AN, Hansbro PM. Harnessing regulatory T cells to suppress asThma: from potential to Therapy. Am J Respir Cell Mol Biol 2010; 43:511-9.
55.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: acommitted regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A. 2009; 106:1903-8.
56.Zhang H, Xia N, Tang T, Nie S, Zha L, Zhang M, Lv B, Lu Y, Jiao J, Li J, Cheng X. Cholesterol suppresses human iTreg differentiation and nTreg function through mitochondria-related mechanisms. J Transl Med. 2023; 21:224.
57.Zhao ST, Wang CZ. Regulatory T cells and asthma. J Zhejiang Univ Sci B. 2018; 19:663-673.
58.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol 2001; 2:301-6.
59.Thorburn AN, Hansbro PM. Harnessing regulatory T cells to suppress asthma: from potential to therapy. Am J Respir Cell Mol Biol 2010; 43:5119.
60.Turner JA, Stephen-Victor E, Wang S, et al. Regulatory T cell-derived TGF-beta1 controls multiple checkpoints governing allergy and autoimmunity. Immunity 2020; 53:12021214.e1206.
61.Seitz C, Liu S, Klocke K, Joly AL, Czarnewski PV, Tibbitt CA, Parigi SM, Westerberg LS, Coquet JM, Villablanca EJ, Wing K, Andersson J. Multi-faceted inhibition of dendritic cell function by CD4 + Foxp3 + regulatory T cells. J Autoimmun 2019; 98:86-94.
62.Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, Flores M, Li N, Schweighoffer E, Greenberg S, Tybulewicz V, Vignali D, Clynes R. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol 2008; 180:5916-26.
63.Garín MI, Chu CC, Golshayan D, Cernuda-Morollón E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood 2007; 109:2058-65.
64.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 2012; 12:180-90.
65.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 2004; 4:665-74.
66.de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol 2004; 34:2480-8.
67.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant Th2-like bronchoalveolar T-lymphocyte population in atopic asThma. N Engl J Med 1992; 326:298-304.
68.Kim HY, DeKruyff RH, Umetsu DT. The many paThs to asThma: phenotype shaped by innate and adaptive immunity. Nat Immunol 2010; 11:577-84.
69.Johnston RA, Zhu M, Rivera-Sanchez YM, Lu FL, Theman TA, Flynt L, Shore SA. Allergic airway responses in obese mice. Am J Respir Crit Care Med 2007; 176:650-8.
70.Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, Zhu M, Iwakura Y, Savage PB, DeKruyff RH, Shore SA, Umetsu DT. Ozone exposure in a mouse model induces airway hyperreactivity That requires The presence of natural killer T cells and IL-17. J Exp Med 2008; 205:385-93.
71.Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, Benoit LA, Byers DE, Alevy Y, Tucker J, Swanson S, Tidwell R, Tyner JW, Morton JD, Castro M, Polineni D, Patterson GA, Schwendener RA, Allard JD, Peltz G, Holtzman MJ. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med 2008; 14:633-40.
72.Wright RJ. Stress and atopic disorders. J Allergy Clin Immunol 2005; 116:1301-6.
73.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie AN. Nuocytes represent a new innate effector leukocyte That mediates type-2 immunity. Nature 2010; 464:1367-70.
74.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H. Innate Lymphoid Cells: 10 Years On. Cell 2018; 174:1054-1066.
75.Yu, S, Kim, HY, Chang, YJ, DeKruyff, RU, Umetsu DT. Innate lymphoid cells and asThma.JACI 2014; 133:943-950.
76.Ebbo M, Crinier A, Vély F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol 2017; 17:665-678. doi: 10.1038/nri.2017.86.
77.Licona-Limón P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol 2013; 14:536-42.
78.Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, Voehringer D, Busslinger M, Diefenbach A. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012; 37:634-48.
79.Klein Wolterink RG, Serafini N, van Nimwegen M, Vosshenrich CA, de Bruijn MJ, Fonseca Pereira D, Veiga Fernandes H, Hendriks RW, Di Santo JP. Essential, dose-dependent role for The transcription factor Gata3 in The development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc Natl Acad Sci U S A 2013; 110:10240-5.
80.Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, te Velde AA, Fokkens WJ, van Drunen CM, Spits H. The transcription factor GATA3 is essential for The function of human type 2 innate lymphoid cells. Immunity 2012; 37:649-59.
81.Helfrich S, Mindt BC, Fritz JH, Duerr CU. Group 2 Innate Lymphoid Cells in Respiratory Allergic Inflammation. Front Immunol 2019; 10:930.
82.Barlow JL, McKenzie AN. Type-2 innate lymphoid cells in human allergic disease. Curr Opin Allergy Clin Immunol 2014; 14:397-403.
83.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates Th2 cytokine production. J Allergy Clin Immunol. 2013 Jul; 132(1):205-13.
84.Antosz K, Batko J, Blazejewska M, Gawor A, Sleziak J, Gomulka K. Insight into IL-5 as a Potential Target for the Treatment of Allergic Diseases. Biomedicines 2024; 12:1531.
85.McBrien CN, Menzies-Gow A. The Biology of Eosinophils and Their Role in Asthma. Front Med (Lausanne) 2017; 4:93.
86.Sehmi R, Wardlaw AJ, Cromwell O, Kurihara K, Waltmann P, Kay AB. Interleukin-5 selectively enhances the chemotactic response of eosinophils obtained from normal but not eosinophilic subjects. Blood 1992; 79:2952-9.
87.Johnson JR, Wiley RE, Fattouh R, Swirski FK, Gajewska BU, Coyle AJ, Gutierrez-Ramos JC, Ellis R, Inman MD, Jordana M. Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am J Respir Crit Care Med 2004; 169:378-85.
88.Ma J, Rubin BK, Voynow JA. Mucins, Mucus, and Goblet Cells. Chest 2018; 154:169-176.
89.Rogers DF. Airway goblet cell hyperplasia in asthma: hypersecretory and anti-inflammatory? Clin Exp Allergy 2002; 32:1124-7.
90.Xue L, Salimi M, Panse I, Mjösberg JM, McKenzie AN, Spits H, Klenerman P, Ogg G. Prostaglandin D2 activates group 2 innate lymphoid cells Through chemoattractant receptor-homologous molecule expressed on Th2 cells. J Allergy Clin Immunol 2014; 133:1184-94.
91.Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, Wechsler ME, Israel E, Levy BD. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asThma. Sci Transl Med. 2013; 5:174ra26. doi: 10.1126/scitranslmed.3004812.
92.Maric J, Ravindran A, Mazzurana L, Van Acker A, Rao A, Kokkinou E, Ekoff M, Thomas D, Fauland A, Nilsson G, Wheelock CE, Dahlén SE, Ferreirós N, Geisslinger G, Friberg D, Heinemann A, Konya V, Mjösberg J. Cytokine-induced endogenous production of prostaglandin D 2 is essential for human group 2 innate lymphoid cell activation. J Allergy Clin Immunol 2019; 143:2202-2214.e5.
93.van Rijt L, von RichThofen H, van Ree R. Type 2 innate lymphoid cells: at The cross-roads in allergic asThma. Semin ImmunopaThol 2016; 38:483-96.
94.Goswami R, Kaplan MH. A brief history of IL-9. J Immunol 2011; 186:3283-8.
95.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, SaThaliyawala T, Kubota M, Turner D, Diamond JM, GoldraTh AW, Farber DL, Collman RG, Wherry EJ, Artis D. Innate lymphoid cells promote lung-tissue homeostasis after infection wiTh influenza virus. Nat Immunol 2011; 12:1045-54.
96.Zaiss DMW, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015; 42:216-226.
97.Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, Fahy RJ, Crotty TB, Hirani N, Flynn RJ, Voehringer D, McKenzie AN, Donnelly SC, Fallon PG. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A.2014; 111:367372.
98.Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, Spits H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTh2 and CD161. Nat. Immunol 2011; 12:10551062.
99.Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, HepworTh MR, Van Voorhees AS, Comeau MR, Artis D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med 2013; 5(170):170ra16.
100.Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST, McKenzie AN, Fallon PG, Ogg GS. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 2013; 210:29392950.
101.Doherty TA, Scott D, Walford HH, Khorram N, Lund S, Baum R, Chang J, RosenThal P, Beppu A, Miller M, Broide DH. Allergen challenge in allergic rhinitis rapidly induces increased peripheral blood type 2 innate lymphoid cells That express CD84. J Allergy Clin Immunol 2014;133:12031205.
102.Beale J, Jayaraman A, Jackson DJ, Macintyre JDR, Edwards MR, Walton RP, Zhu J, Man Ching Y, Shamji B, Edwards M, Westwick J, Cousins DJ, Yi Hwang Y, McKenzie A, Johnston SL, Bartlett NW. Rhinovirus-induced IL-25 in asThma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Trans Med 2014;6:256ra134. doi: 10.1126/scitranslmed.3009124. PMID: 25273095; PMCID: PMC4246061.
103.Hong JY, Bentley JK, Chung Y, Lei J, Steenrod JM, Chen Q, Sajjan US, Hershenson MB. Neonatal rhinovirus induces mucous metaplasia and airways hyperresponsiveness Through IL-25 and type 2 innate lymphoid cells. J Allergy Clin Immunol 2014;134:429439.
|
|
Home
Design by Walter Serralheiro
|
|
|