Asma Brônquica Tratamento da Asma Mecanismo de Ação dos Corticoides Os corticoides desempenham um papel vital nas funções e nos mecanismos homeostáticos que governam a função fisiológica normal da celula. Suas funções no organismo são múltiplas, incluindo atuação no metabolismo de carboidratos, proteínas e lípides, manutenção do equilíbrio hidro-eletrolítico, bem como na preservação da função normal cardiovascular, rins, músculos, sistema endócrino e sistema nervoso. Até há pouco tempo os efeitos dos corticoides eram vistos como fisiológicos (refletindo as ações do cortisol produzido diariamente pelas adrenais) ou patológicos (síndrome de Cushing e insuficiência suprarrenal). Posteriormente surgiram os conceitos de anti-inflamação e imunossupressão, que tornaram os corticoides uma das classes terapêuticas mais importantes da atualidade, com múltiplas indicações em doenças inflamatórias como artrite reumatoide, doenças inflamatórias intestinais e doenças auto-imunes, tornando-se inclusive o principal tratamento para a inflamação alérgica, incluindo a da asma brônquica. Na asma, os conhecimentos sobre a patologia celular e molecular da inflamação brônquica só têm aumentado, demonstrando que existem múltiplos alvos para a ação dos corticoides in vivo. O melhor entendimento dos mecanismos fundamentais da gene transcrição determinaram grandes progressos na compreensão dos mecanismos moleculares pelos quais os corticoides suprimem a inflamação. A inflamação na asma é caracterizada pelo aumento da expressão de múltiplos genes inflamatórios, como os que codificam as citocinas, quimocinas, moléculas de adesão, enzimas inflamatórias e receptores. O aumento da expressão destes genes é regulado por fatores de transcrição, como o fator nuclear-kappa B (NF-kB) e ativador proteico-1 (AP-1).1,2 Estes se ligam e ativam moléculas coativadoras, as quais acetilam o núcleo das histonas e ativam a gene transcrição. Os corticoides atuam suprimindo múltiplos genes pela reversão da acetilação da histona. Agem através da ligação aos receptores ativados de corticoides, a coativadores e pelo recrutamento de histona deacetilases (HDACs) para o complexo de transcrição ativado. Inibem os fatores de transcrição que regulam a gene expressão anormal. Também aumentam a transcrição de genes que codificam proteínas anti-inflamatórias, como lipocortin-1, IL-10, antagonista do receptor IL-1 e a endopeptidase neutra. Para se entender como os corticoides operam na asma torna-se necessário compreender as bases moleculares da inflamação, o remodelamento da cromatina, como funcionam os Receptores de Glicocorticoides (GR), a gene transcrição corticoide-induzida, a interação do corticoide com os fatores de transcrição e os efeitos da acetilação das histonas. Como a inflamação foi já descrita nos capítulos sobre a Resposta Tardia da Asma, serão aqui descritos de forma sucinta os outros tópicos. Remodelamento da Cromatina O DNA é o responsável pelo armazenamento e transmissão da informação genética. Um gene é um segmento de DNA que contém o arquivo completo da sequência de aminoácidos para fabricar uma cadeia polipeptídica específica. O DNA é encontrado principalmente nos cromossomos e, em pequenas quantidades, nas mitocôndrias. O DNA de uma célula humana apresenta um comprimento total de quase 2 metros. Provavelmente, para facilitar a organização desses quase 2 metros dentro do núcleo de cada célula, o DNA é dividido em vários elementos distintos chamados cromossomos. Cada cromossomo é formado por uma única molécula de dupla hélice de DNA condensada com proteínas. O DNA de todos os cromossomos encontra-se "empacotado" em estrutura densa, com o auxílio de proteínas básicas especializadas. Estas proteínas básicas que se ligam ao DNA são divididas em duas classes gerais: as histonas e as proteínas cromossômicas não-histônicas. O complexo formado pelo DNA e proteína é chamado de nucleossomo e compõe uma fibra com aspecto de contas num colar. Como vez por outra, por exemplo, quando um gene é ativado, a molécula de DNA deve abrir num ponto específico para permitir que o RNA copie sua mensagem, é fácil imaginar que o empacotamento do DNA nos cromossomos deve seguir um esquema muito rigoroso para não comprometer a molécula. O complexo formado pela interação de proteínas de ambas as classes com o DNA nuclear é conhecido como cromatina (o material do qual os cromossomos são feitos). As histonas estão presentes numa quantidade tão grande (em torno de 60 milhões de moléculas de cada tipo de histona por célula) que a sua massa total de cromatina é aproximadamente igual àquela do DNA. As histonas têm um papel fundamental na compactação, de uma maneira organizada, das moléculas de DNA muito longas, em um núcleo que possui apenas uns poucos micrômeros de diâmetro. Existem cinco tipos de histonas em dois grupos principais: as histonas nucleossômicas (H2A, H2B, H3 e H4) e os da H1. As histonas nucleossômicas são pequenas proteínas (102-135 aminoácidos) responsáveis pelo enrolamento do DNA nos nucleossomos. O nucleossomo, subunidade fundamental da cromatina, consiste de aproximadamente 146 bp de DNA. É formado por um núcleo octâmero composto por oito subunidades com duas cópias de de cada histona, 2A, 2B, 3 e 4 em volta do qual o DNA é enrolado duas vezes. A facilidade com que um segmento de DNA curva-se para dar essas duas voltas em torno de um nucleossomo varia em função da sua sequência nucleotídica.3 A consequência funcional de empacotar a cromatina é a de limitar o acesso de fatores da transcrição ao DNA. A cromatina quando visualizada ao microscópio pode apresentar-se opaca ou clara devido ao enrolamento ou não do DNA ao redor do núcleo da histona.4 As histonas são proteínas com uma proporção muito grande de aminoácidos positivamente carregados (lisina e arginina). As cargas positivas ajudam as histonas a se ligarem fortemente ao DNA (que tem um alto conteúdo de cargas negativas), de uma forma independente da sequência nucleotídica.4 Cada núcleo de histona possui uma longa zona terminal rica em resíduos de lisina que podem ser acetilados, modificando a carga elétrica do núcleo da histona. Na célula "em repouso" o DNA está fortemente enrolado, impedindo a ligação da RNA polimerase II (Pol II), que ativa a formação do RNA mensageiro (mRNA). Na realidade, a modificação das histonas pela acetilação ou metilação modifica suas cargas e afeta o enrolamento (empacotamento) do DNA.
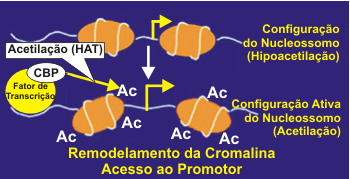
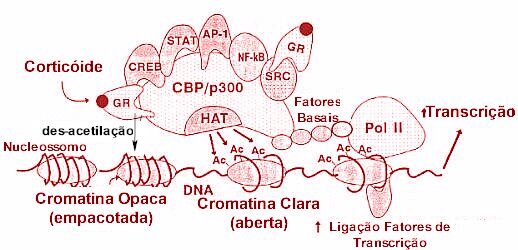
A deacetilação da histona aumenta o enrolamento do DNA ao redor dos resíduos de histona, resultando em cromatina mais densa, o que determina um acesso restrito aos fatores de transcrição aos seus sítios de ligação, e desse modo reprime a gene transcrição.5 Alterações na atividade HAT e HDAC podem influenciar as doenças inflamatórias crônicas. Em estudos recentes, fragmentos de biópsias brônquicas de pacientes com asma evidenciam expressão de altos níveis de atividade HAT e baixa atividade HDAC, sugerindo que um desequilíbrio HAT-HDAC possa ser o substrato para a gene expressão pró-inflamatória da doença. Quando os fatores de transcrição pró-inflamatórios envolvidos na patogênese da asma, como o NF-kB, o AP-1 e o NF-AT são ativados, eles se ligam a sequências específicas de reconhecimento no DNA e posteriormente interagem com moléculas coativadoras como a p/300/CREB (cyclic adenosine monophsophate response element-binding protein)-binding protein (CBP) e p300/CBP-associated factor (PCAF). Estas moleculas coativadoras são proteínas nucleares, atuam como um "interruptor" molecular que controla a gene transcrição, via atividade intrínseca histona acetiltransferase, resultando na acetilação dos núcleos da histona, reduzindo a sua carga.6,7 A acetilação determina a transformação da cromatina condensada em uma forma ativada aberta. A gene transcrição com o DNA ocorre após o remodelamento, com a estrutura da cromatina aberta, permitindo que os complexos de transcrição possam se ligar ao DNA e iniciar a transcrição, formando o RNA mensageiro (mRNA) e síntese de proteíinas inflamatórias que alteram a função celular (Figura 2).
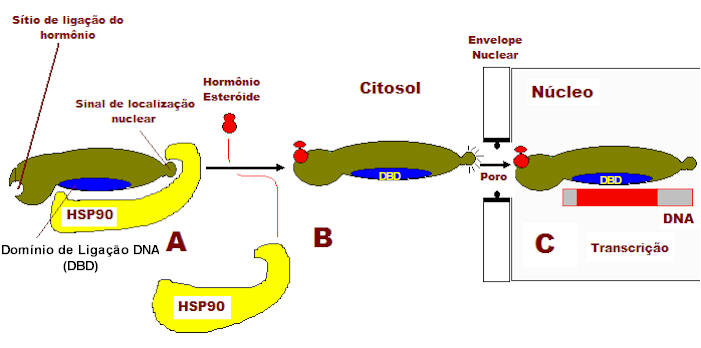
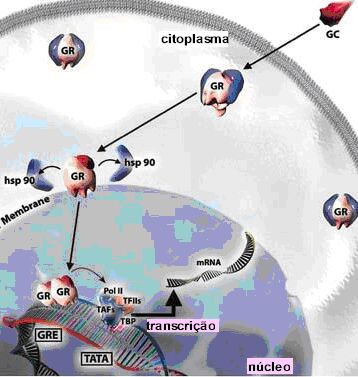
Os corticoides atuam como anti-inflamatório através da interação com os GR que se encontram no citoplasma das células e que após ativação migram para o núcleo da célula onde intervêm com a maquinária de transcição junto ao DNA, recrutam enzimas especializadas na acetilação e remodelamento da cromatina, e na geração ou supressão da produção de mRNAs de grande número de substâncias que influenciam a inflamação da asma. Receptor de Glicocorticoide Os receptores nucleares constituem uma das classes mais ricas em reguladores transcricionais nos animais (metazoários). Eles regulam diversas funções, como homeostase, desenvolvimento, reprodução, metabolismo e podem ser importantes em doenças crônicas (inflamatórias, imunológicas e neoplásicas). Existem mais de 100 receptores nucleares conhecidos, que juntos constituem a superfamília dos receptores nucleares, da qual o receptor de esteroide glicocorticoide (GR, NR3C1) é um de seus membros.8 Pelo menos 49 genes já foram identificados, sendo que o gene do GR está localizado no cromossomo 5, na região 5q31,9 embora outras diferentes variantes sejam agora reconhecidas.10 A estrutura genômica do gene GR consiste de 9 éxons estendendo-se por mais de 80 kb do genoma humano, com codificação proteica iniciando-se no éxon 2. O receptor consiste de domínios distintos, incluindo o domínio N-terminal (NTD), o domínio de ligação de DNA central e o domínio de ligação do ligante C-terminal (LBD) [ 1 , 2 ]. Na ausência de ligante, o GR está localizado no citoplasma em um complexo multiproteico contendo proteínas de choque térmico e outros chaperones. 3 Após a ligação do ligante, o GR é liberado desse complexo e translocado para o núcleo. No núcleo, ele se liga ao DNA através de sequências específicas de DNA (GRE - elemento de resposta glicocorticoide) e regula a transcrição dos genes alvo [ 2 , 4 , 5 ]. O domínio LBD contém uma função de ativação dependente de ligante (AF-2), cuja alteração conformacional na ligação do agonista estabiliza o receptor em uma conformação ativa, facilitando sua interação com coativadores através dos motivos LXXLL [ 6 , 7 ]. Os fatores de transcrição são proteínas que se ligam a sequências controladoras, geralmente na 5' upstream da região promotor de genes alvo, para aumentar (e às vezes diminuir) a taxa de gene transcrição, resultando em aumento ou redução na síntese de proteínas e subsequente alteração da função celular. Os fatores de transcrição podem interagir uns com os outros, resultando em inibição e as vezes em aumento da atividade transcricional. Na asma, o excesso de ativação dos fatores de transcrição (NF-kB, AP-1 e NF-AT) pode ser responsável por prolongada liberação de mediadores da inflamação. Várias são as subclasses de fatores de transcrição e os membros de cada família compartilham características estruturais. Estas famílias incluem a hélice-volta-hélice (helix-turn-helix) (p.ex. POU), dedos de zinco (p.ex. esteroides), Zíper de leucina (p.ex. CREB, NF-kB, AP-1) e folha beta (beta-sheet motifs) (p. ex. HU). Muitos fatores de transcrição são comuns a vários tipos de células (ubíquos) e podem atuar na regulação de genes inflamatórios, enquanto que outros são específicos para certas células, podendo determinar as suas características fenotípicas. A superfamília dos receptores nucleares incluem receptores cujos ligantes já foram identificados, além de outros, em maior número, que não possuem ligantes conhecidos, chamados de receptores órfãos. Os receptores para os hormônios esteroides formam uma subclasse da superfamília de receptores nucleares. Na ausência de ligante, os receptores dos esteroides glicocorticoides, mineralocorticoides, androgênios, estrogênio e progesterona estão associados em um complexo com proteínas de choque térmico (HSPs) no citoplasma e, em alguns casos, no núcleo da célula. A ligação do hormônio dissocia os receptores deste complexo e induz a formação de homodímeros que se dirigem ao núcleo onde se ligam a sequências específicas no DNA denominadas elementos responsivos ao hormônio (HRE), regulando a transcrição. Estes hormônios são pequenas moléculas hidrofóbicas que diferem bastante umas das outras em estrutura química e função. No entanto, todas atuam através de mecanismos similares, como o corticoide utilizado na terapêutica da asma. São moléculas que se difundem passivamente diretamente através da membrana plasmática das células-alvo e ligam-se a receptores protéicos intracelulares, ativando-os. O acoplamento do ligante ativa receptores, os quais regulam a transcrição de genes específicos. O GR e outros receptores hormonais desta família estão estruturalmente organizados em cinco domínios homólogos funcionais, cada um responsável por certas funções11 (Figura 3): 1. Domínio NT (amino-terminal) - domínio de transativação; 2. Domínio de ligação ao DNA (DBD - DNA-binding domain); 3. Região de dimerização do receptor de corticoide; 4. Região CT (carboxi-terminal) com o domínio de ligação ao ligante (LBD - ligant binding domain); 5. Regiões de localização nuclear.
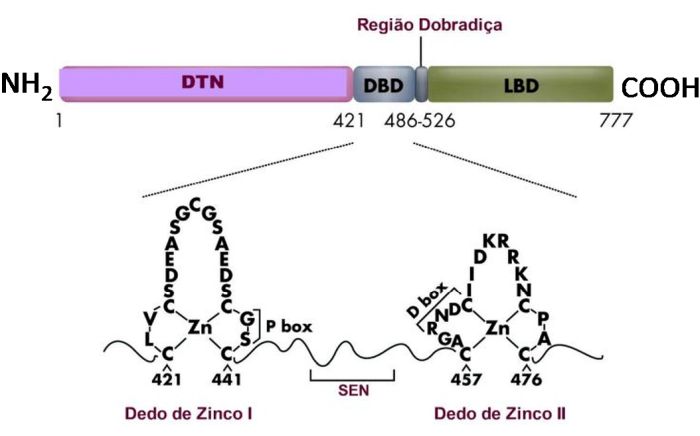
A representação linear do GR é constituída por 777 aminoácidos. O domínio amino terminal (NT) é composto por aproximadamente 439 aminoácidos e é o principal responsável pela maior parte da atividade transcricional independente do ligante, denominada função de ativação 1 (AF-1). É o sítio imunogenético do receptor e também possui importante especificidade junto ao promotor. O GR é fosforilado predominantemente nos resíduos serina do NT porém, o papel da fosforilação na ação dos corticoides ainda não está elucidada. Sítios de fosforilação ocorrem dentro da região de transativação correspondendo ao AF-1. A região AF-1 contém 200 aminoácidos, com função de ativação transcricional hormônio-independente.12 A atividade transcricional do AF-1 parece residir na região central de 41 aminoácidos carregados negativamente por aminoácidos ácidos.13 Tem-se demonstrado que esta região interage com fatores gerais de transcrição e com um número de proteínas coativadoras, apresentando ainda, importante participação na repressão transcricional pelo GR (região AF-1). Os corticoides são extremamente lipofílicos e por difusão passiva penetram diretamente através da membrana plasmática das células-alvo no citoplasma e ligam-se aos GRs intracelulares em um sítio conhecido como domínio de ligação do hormônio (LBD - ligant binding domain) ativando-os, determinando uma mudança na configuração do receptor. Este domínio localiza-se na região carboxi-terminal (CT) e possui várias funções, a homo e heterodimerização do receptor, localização nuclear, dissociação das proteínas de choque HSP90 (heat shock protein 90) e interação com proteínas correpressoras e coativadoras. Além dessas funções, o LDB contém uma superfície que é fundamental para a ativação transcricional, que se forma com a ligação do hormônio ao receptor. O domínio CT primariamente controla a atividade do receptor como um todo através de sua interação com as HSPs90 e ciclofilinas (quando o receptor está inativo) ou com hormônios e coativadores (quando o receptor está ativo). É o local onde ocorre a ligação da proteína de choque térmico que atua como molécula chaperone (acompanhante, em inglês), que é a responsável em parte, na manutenção da conformação inativa do GR, encobrindo os sinais de localização nuclear localizados nessa região.11 Sem ligante, o GR encontra-se predominantemente no citosol como polímero de baixo peso molecular (aproximadamente 90 kd), parte de um complexo multiprotéico inativo (aproximadamente 330 kd), que consiste de duas moléculas HSP90, e outras proteínas como, p59, p60, p23, imunofilinas e calreticulinas.14 As HSPs90 associadas ao GR parecem ser importantes na manutenção do GR em uma conformação que seja apropriada para a ligação/ligante e para a inibição da translocação do GR para o núcleo. A ligação GR-HSP não permite a dimerização, a ativação e a ligação do complexo receptor-esteroide ao DNA. O transporte do GR para o núcleo da célula é mediado por duas sequências de localização nuclear (NLSs), NL1 e NL2. A NL2 está dentro do LBD e medeia uma transferência parcial do GR para o núcleo em vários tipos de células, enquanto que o NL1 é um motivo básico curto no domínio dobradiça do GR (entre DBD (DNA binding domain) e LBD) que é suficiente para a completa transferência nuclear do receptor. Esta região é também o local de rotação das hélices do receptor, indispensável para permitir a alteração conformacional do complexo receptor-corticoide e é responsável pela especificidade do gene alvo, bem como pela alta afinidade de ligação ao DNA.
Estas estruturas em dedo de zinco conferem a alta afinidade dos GRs aos elementos responsivos ao corticoide (GREs) nas regiões promotoras do DNA.11 Os GREs são sequências específicas de DNA na região promotor de genes implicados na asma, onde o GR se liga e promove a "responsividade" (mensagem específica) glicocorticoide aos genes (como no caso dos B2-receptores).15 Por outro lado, o corticoide pode promover uma regulação negativa da transcrição (transrepressão), via GRE negativo (nGRE) (como no caso da osteocalcina).15 Acredita-se que todos os receptores proteicos intracelulares fixam-se ao DNA como homodímeros ou heterodímeros.
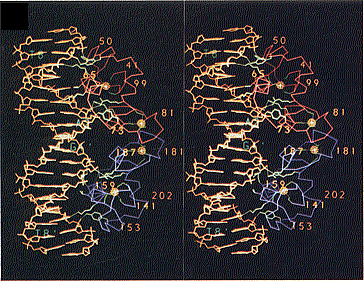
A Figura 6 apresenta fotografia estereoscópica do GRE com o GR.
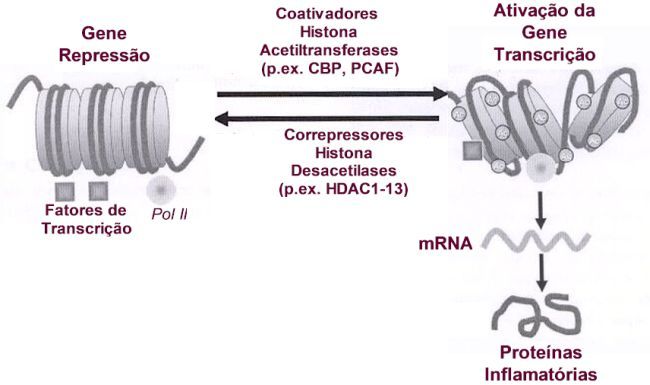
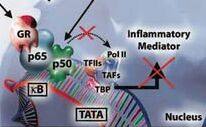
O homodímero GR interage com o complexo basal de transcrição junto a caixa TATA (TATA box). O complexo basal de iniciação da transcrição inclui a proteína que se liga a TATA (TBP) assim como a RNA polimerase II que requer a ajuda de um grande conjunto de fatores gerais de transcrição (TAFs e TFIIs). Os fatores gerais de transcrição são responsáveis pelo posicionamento correto da enzima RNA polimerase II no promotor. Ajudam na separação das fitas de DNA para permitir o início da transcrição e liberam a RNA polimerase do promotor quando a transcrição se inicia. A interação entre o GR e o complexo basal de transcrição otimiza a transcrição de genes alvo (Figura 7). Coreguladores Enquanto o papel de fatores gerais de transcrição na mediação da transcrição basal já está bem documentado, ficou estabelecido que os receptores nucleares recrutam fatores auxiliares chamados de coreguladores: coativadores (proteínas nucleares que ativam a gene transcrição via atividade intrínseca histona acetiltransferase) e corepressores (proteínas nucleares que reprimem a gene transcrição e tem atividade histona desacetilase). Estes criam, dependendo do estado de ativação do receptor, uma ação permissiva (estimulação) ou repressora de transcrição junto ao promotor, e se comunicam com os fatores gerais de transcrição e com a RNA Polimerase II.8 As moléculas proteicas coativadoras modulam a atividade do GR e podem apresentar um papel na especificidade celular dos efeitos do GR. Formam interações com o GR e a maquinária basal de transcrição, servindo como elo de ligação entre DNA associado ao GR e os fatores gerais de transcrição que aumentam a trancrição. Vários grupos distintos de proteínas coativadoras têm sido identificados, com atuação na ativação transcricional mediada pelo GR.16 Estes coativadores constituem subunidades de grandes complexos multiprotéicos que atuam em vários níveis funcionais, remodelando a cromatina, determinando modificação enzimática das histonas ou modulação do complexo de pré-iniciação via interação com a Pol II e GTFs. A ligação dos receptores nucleares aos coativadores é frequentemente mediada por seus domínios de ativação transcricional. Os domínios AF-1 e AF-2 parecem ser os sítios de contato para o GR com coativadores, formando complexos protéicos envolvidos na ativação da transcrição. A proteína de ligação CBP (cAMP response element binding protein (CREB)- binding protein) é um coativador nuclear ubíquo que funciona, como integrador da gene transcrição, unindo ativadores de transcrição, tais como o GR, com o complexo basal de iniciação da transcrição. A CBP foi descoberta primeiramente como um coativador da CREB.17 Uma outra proteína relacionada, a chamada p300, apresenta homologia com a CBP. Ambas têm demonstrado formar interações proteína-proteína com membros do dispositivo basal transcricional incluindo a RNA polimerase II (Pol II) e fatores gerais de transcrição TFIIB (transcription factor II B) a proteína que se liga a TATA (TBP). Posteriormente ficou demonstrado ligarem-se a vários outros fatores de transcrição, como o AP-1 e NF-kB que upregulate vários genes inflamatórios da asma. Quando os fatores de transcrição pró-inflamatórios NF-kB e AP-1 são ativados, eles se ligam a sequências específicas de reconhecimento no DNA e posteriormente também interagem com moléculas coativadoras como a p300/CBP e p300/CBP-associated factor (PCAF). Estas moleculas coativadoras atuam como um "interruptor" molecular que passa a controlar a gene transcrição. Todos têm atividade intrínseca de acetilação da histona, que resulta na acetilação dos núcleos da histona, reduzindo a sua carga. Outras proteínas coativadoras do GR incluem as da família SRC (steroid receptor coactivator), que consiste em três novos coativadores identificados: Src-1 (12), GRIP1 (glucocorticoid receptor-interacting protein 1),8,18,19 e p/CIP.20,21 Os GR após ativação pelos corticoides podem ligar-se aos coativadores SRC que se ligam a CBP, resultando em aumento da transcrição. SRC e CBP apresentam atividade intrínseca HAT e são capazes de acetilar histonas. Resultados de análise in vitro sugerem que os SRCs medeiam a atividade transcricional através de múltiplos mecanismos, incluindo: 1. A interação direta com receptores nucleares ligant-bound;8 2. Contato direto com determinados fatores gerais de transcrição, tais como TFIIB e TBP;22 3. Interação com os coativadores transcricionais comuns, tais como CBP, p300, e PCAF;8 4. Interação com outros coativadores tais como o CARM-1 (coactivator-associated arginine methyltransferase 1), ASC-2 (cancer-amplified transcriptional coactivator), PGC-1 (PPARg coactivator-1) e o SRA (steroid receptor RNA co-activator);23-25 5. Participação no remodelamento da cromatina através de sua atividade intrínseca da acetiltransferase da histona (HAT), que altera a conformação de nucleossomos na região promotor e permite a ligação de fatores necessários para aumentar a transcrição;26-28 e 6. Modificação enzimática de outros constituintes do complexo coativador.29 A ligação do GR ao dispositivo basal de transcrição não é a única função das proteínas coativadoras, que atuam na gene transcrição modulando a estrutura da cromatina. CDP-p300 e SRC-1 apresentam atividade HAT, alterando a conformação dos nucleossomos, que influencia o acesso ao promotor de fatores de transcrição, aumentando a transcrição. Em contraposição, têm sido descritas proteínas com atividade HDAC, que reduzem a acetilação da histona e restauram a estrutura densa da cromatina, o que inibe a ligação de fatores de transcrição. Receptores ativados de glicocorticoides podem ligar-se diretamente a CBP ou outros coativadores para inibir a atividade HAT, revertendo o desenrolar do ADN ao redor do núcleo da histona, remodelando a cromatina e assim reprimir os genes inflamatórios. Baixas concentrações de corticoides ativam os receptores de corticoides, recrutam HDACs (principalmente HDAC1 e 2) para ativar o complexo transcricional, resultando na desacetilação das histonas, reduzindo a transcrição de genes inflamatórios e aumentam a transcrição de genes anti-inflamatórios. Ao menos 11 HDACs já foram identificadas em mamíferos e são agrupadas em duas classes. A classe I engloba HDAC1, 2, 3, 8 e 11, as quais apresentam significante homologia com a proteína RPD3 (um regulador transcricional, e na levedura uma conhecida histona desacetilase) e estão localizadas preferencialmente no núcleo. A classe II inclui as HDAC4, 5, 6, 7, 9 e 10, as quais são homólogas as enzimas HAD1-like da levedura e transitam indo e vindo entre o núcleo e o citoplasma. As HDACs da classe I são largamente expressas na maioria das células, enquanto que as da classe II são mais restritas, podendo estar envolvidas na diferenciação celular. Estas diferenças nas HDACs podem contribuir para diferenças na responsividade aos corticoides entre genes e células distintos. A ativação de genes anti-inflamatórios pelos corticoides está associada a acetilação seletiva dos resíduos de lisina 5 e 16 na histona-4, resultando aumento na gene transcrição.30,31
Uma questão importante é por que os corticoides desativam apenas os genes inflamatórios: eles nitidamente não suprimem a atividade de todos os genes e são bem tolerados como terapêutica. Os receptores de corticoides provavelmente ligam-se apenas aos coativadores que são ativados por fatores de transcrição pró-inflamatórios, como o NF-kB e AP-1, embora não se saiba como ocorre esta ação específica de reconhecimento. Um conceito emergente sugere que os ativadores da transcricão, como o GR, devam agir também em uma outra enzima que atue no remodelamento da cromatina, além dos coativadores com atividade HAT. Estes complexos remodeladores cromatina ATP-dependentes são conhecidos como Swi/Snf (switch/sucrose nonfermentable). O complexo Swi/Snf hidrolisa o ATP e utiliza a energia da hidrólise do ATP para romper interações histona-DNA e remodelar a cromatina, permitindo que os fatores da transcrição se liguem ao promotor.32 Embora o exato mecanismo pelo qual o Swi/Snf altera a estrutura do cromatina não esteja de todo elucidado, já existem evidências de que o recrutamento do Swi/Snf é essencial para a regulação da transcrição do gene, incluindo aqueles mediados pelo GR.33
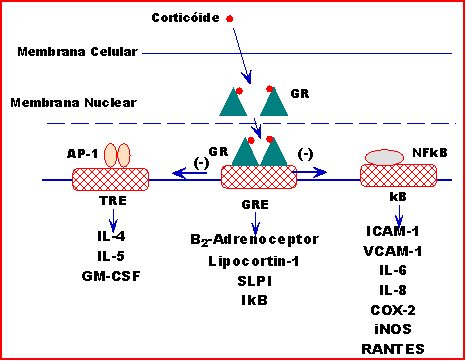
■ Por controle direto ou indireto sobre a transcrição de certos genes-alvo.34,36 O número de genes controlados diretamente pelo corticoide por célula é estimado entre 10 e 100, sendo que outros são regulados indiretamente através de interação com diferentes fatores de transcrição (Figura 9). Os corticoides downregulate importantes citocinas e quimocinas implicadas na asma, interferindo com a ligação dos fatores de transcrição pró-inflamatórios com seus sítios cognatos de ligação DNA. ■ Por interação direta proteína-proteína com o fator de transcrição AP-1 e o GR.37 Funcionalmente, as consequências destas interações resultam em uma mútua repressão transcricional tanto do AP-1 como do GR38,39 o que inibe os efeitos pró-inflamatórios de uma série de citocinas.
■ Por aumento das ribonucleases das células, e desta forma reduzindo os níveis de mRNA.45 ■ As maiores evidências sugerem que a regulação dos genes-alvo ocorre ao nível da transcrição, entretanto, em algumas citocinas a regulação da gene expressão é, ao menos parcialmente, postranscricional.46,47 A repressão dos genes que codificam a IL-11, o GM-CSF e a ciclooxigenase nas células epiteliais do pulmão e fibroblastos é mediada por mecanismos transcricional e postranscricional.48-50 ■ Os corticoides apresentam também ações anti-inflamatórias não ligadas a transcrição através do remodelamento da cromatina. Uma delas é o reconhecimento de sua participação interferindo na síntese de proteínas pela redução da estabilidade do mRNA, reduzindo a síntese protéica. Alguns genes inflamatórios, como o que codifica o GM-CSF, produzem mRNA que são particularmente susceptíveis a ação de ribronucleases que degradam o mRNA, e desta forma interrompem a síntese de proteínas. Os corticoides podem ter efeitos inibitórios nas proteínas que estabilizam mRNA, determinando uma degradação mais rápida e desta forma uma redução na expressão da proteína.51 ■ Existem evidências de que os corticoides exercem ação inibitória sobre a mitogen-actived protein (MAP) quinases, que apresentam importante participação na gene expressão através da regulação de fatores de transcrição pró-inflamatórios. Os corticoides podem inibir a AP-1 e NF-kB via um efeito inibitório na c-jun N-terminal quinases, a qual ativa estes fatores de transcrição.52,53 Os corticoides reduzem a estabilidade do mRNA para alguns genes inflamatórios, como para ciclooxigenase-2, através de ação inibidora em outra MAP quinase, a p38 MAP quinase.54 Este efeito é mediado pela indução de um potente inbidor endógeno p38 MAP quinase denominado MAP quinase fosfatase-1.55 Corticoides constituem a terapêutica mais eficaz da atualidade no controle a longo prazo da asma, reduzindo a inflamação nas vias aéreas pela interação com fatores de transcrição que regulam a expressão gênica, aumentando ou reduzindo a transcrição. Sua ação alcança mais de 30 alvos diferentes: Aumentando a Transcrição: — aumenta (com doses elevadas) a síntese de lipocortin 1 (annexin-1), uma proteína inibidora da fosfolipase A2 da membrana fosfolipídica do ácido araquidônico, que controla a produção de prostaglandinas, PAF e leucotrienos;56,57 — aumenta a transcrição da antiprotease SLPI (secretory leucocyte protease inibitor) nas vias aéreas58 que se opõe às enzimas inflamatórias como a triptase; — aumenta a expressão da endopeptidase neutra (NEP) no epitélio brônquico, degradando taquicininas, reduzindo a inflamação de causa neurogênica;59 — em monócitos e células T, aumenta a gene transcrição do Ik Ba, que se liga no núcleo ao NF-kB e induz a dissociação do NF-kB dos sítios de ligação kB, nos genes alvo; — aumenta a formação da IL-10 (indiretamente) previamente conhecida como CSIF (cytokine-syntesis-inhibitory factor) que inibe o afluxo de eosinófilos e linfócitos T na reação inflamatória tardia pós-provocação alergênica em ratos Brown-Norway;60,61 — aumenta os níveis de IL-12 mRNA que tem papel importante na inibição de síntese inapropriada de IgE;62 — aumenta a formação de antagonista de receptor IL-1 nas células epiteliais brônquicas in vitro e in vivo;63,64 — regulação negativa dos receptores decoy tipo II, que age como antagonista do receptor de IL-1 (IL-1R2).65 — aumenta a expressão dos receptores ß2, dobrando a velocidade de transcrição no pulmão humano, in vitro;66 — potencializa os efeitos dos ß2-agonistas na musculatura lisa brônquica, previne e reverte a taquifilaxia do ß2-receptor das vias aéreas in vivo e in vitro; — aumenta a expressão da proteína Clara cell (CC10, inibidor fososlipase A2);67 — aumenta a síntese da GILZ (glucocorticoid-induced leucine zipper protein) que inibe o NF-kB e AP-1;68,69 — aumento rápido (10 X) na MKP-1 mRNA duas horas após a exposição à droga e um aumento na proteína mitogen-activated protein kinase phosphatase-1 (MKP)-1 em quatro horas, com concomitante inibição da sinalização celular p38 MAP kinase. A MKP-1 é o inibidor endógeno da proinflamatória mitogen-activated protein (MAP) kinase.70 Reduzindo a Transcrição: — limita a expressão de citocinas como IL-1ß , IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-11, IL-13, IL-16, IL-17, IL-18, TNF-a, GM-CSF, SCF (71);71 — limita a expressão de quimocinas IL-8, RANTES, MIP-1a, MCP-1, MCP-3, MCP-4, eotaxina; — limita a expressão de IL-3, IL-5 e GM-CSF pelo eosinófilo, interferindo em sua diferenciação, ativação, quimiotaxia, inibindo sua capacidade de auto-regulação.72-74 O corticoide bloqueia os efeitos destas citocinas, levando à morte celular programada, ou apoptose;75 — inibe a expressão das moléculas de adesão do endotélio e leucócitos, tais como a selectina-E, VCAM-1 e ICAM-1 por ação direta ou por inibição da produção das citocinas IL-1Ã e TNF-ß.76 Esta ação pode diminuir a migração de células inflamatórias para os tecidos; — reduz o número de mastócitos na mucosa das vias aéreas, pela redução na produção de citocinas IgE dependentes, como a IL-3 e GM-CSF, necessárias para a expressão dos mastócitos nas superfícies mucosas;77 — inibe a transcrição dos receptores Nk1 e NK2 e do receptor de bradicinina B2;78 — reduz a produção, das enzimas inflamatórias iNOS (79-81), COX-282,83 e cPLA2 ;84 — inibe a síntese do peptídio endotelina-1, potente broncoconstritor expresso nas células epiteliais de pacientes asmáticos;85,86 — inibe a proliferação de linfócitos T que ocorre em parte por repressão do fator de progressão G1 e da ciclina D3;87 Referências 01.Barnes PJ, Adcok IM. How do corticosteroids work in asthma? Ann Intern Med 2003; 139:359. 02.Barnes PJ, Adcok IM. Transcription factors and asthma. Eur Respir J 1998; 12:221. 03.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. - Biologia Molecular da Célula. 3ª edição. Porto Alegre: Artes Médicas; 1997. 04.Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell 1998; 92:307. 05.Struhl K, Moqtaderi Z. The TAFs in the HAT. Cell 1998; 94:1. 06.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone aceyltransferases. Cell 1996; 87:953. 07.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem 2001; 70:81. 08.McKenna NJ, Rainer BL, O'Malley BE. Nuclear receptor coregulators: cellular and molecular biology. Endocr.Rev 1999; 20:321. 09.Francke U, Foellmer BE. The glucocorticoid receptor gene is in 5q31-q32 [published correction appears in Genomics. 1989; 5:388]. Gemomics 1989; 4:610. 10.Yudt MR, Cidlowski JA. The glucocorticoid receptor: coding a diversity of proteins and responses though a single gene. Mol Endocrinol 2002; 16:1719. 11.Loke TK, Corrigan CJ, Lee TH. Glucocorticoid Effects on Mediator Modulation. In : Jan M. Agosti and Albert L. Sheffer. Biotherapeutic Approaches to Asthma . New York: Marcel Dekker; 2002:327-352. 12.Hollenberg SM, Evans RM. Multiple and cooperative trans-activation domains of human glucocorticoid receptor. Cell 1988; 55:899. 13.Ford J, McEwan IJ, Wright AP, Gustafsson JA. Involvement of the transcription factor IID protein complex in gene activation by N-terminal transactivation domain of glucocorticoid receptor in vivo. Mol Endocrinol 1997; 11:1467. 14.Cheung J, Smith DF. Molecular chaperone interactions with steroid receptors: an update. Mol Endocrinol 2000; 14: 939. 15.Newton R. Molecular mechanisms of glucocorticoid action: What is important? Thorax 2000; 55:603. 16.Featherstone M. Coactivators in transcription initiation: here are your order. Curr Opin Genet Dev 2002; 12:149. 17.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 2001; 114:2363. 18.Hong H, Kohli K, Trivedi A, Johnson DL, Stallcup MR. GRIP1, a novel mouse protein that serves as a trnscriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci USA 1996; 93:4948. 19.Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J 1996; 15:3667. 20.Takeshita A; Cardona GR; Koibuchi N; Suen CS; Chin WW. TRAM-1, A novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid receptor coactivator-1. J Biol Chem 1997; 272:27629. 21.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Resnfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 1997; 387:677. 22.Takeshita A; Yen PM; Misiti S; Cardona GR; Liu Y; Chin WW. Molecular cloning and properties of a full-length putative thyroid hormone receptor coactivator. Endocrinology 1996; 137:3594. 23.Chen D; Ma H; Hong H; Koh SS; Huang SM; Schurter BT; Aswad DW; Stallcup MR. Regulation of transcription by a protein methyltransferase. 24.Lee SK; Anzick SL; Choi JE; Bubendorf L; Guan XY; Jung YK; Kallioniemi OP; Kononen J; Trent JM; Azorsa D; Jhun BH; Cheong JH; Lee YC; Meltzer PS; Lee JW. A nuclear factor, ASC-2, as a cancer-amplified transcriptional coactivator essential for ligand-dependent transactivation by nuclear receptors in vivo. J Biol Chem 1999; 274:34283. 25.Lanz RB; McKenna NJ; Onate SA; Albrecht U; Wong J; Tsai SY; Tsai MJ; O'Malley BW. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999; 97:17. 26.Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell 1998; 92:307. 27.Chen H; Lin RJ; Schiltz RL; Chakravarti D; Nash A; Nagy L; Privalsky ML; Nakatani Y; Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 1997; 90:569. 28. Spencer TE; Jenster G; Burcin MM; Allis CD; Zhou J; Mizzen CA; McKenna NJ; Onate SA; Tsai SY; Tsai MJ; O'Malley BW. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997; 389):194. 29.Chen H; Lin RJ; Xie W; Wilpitz D; Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 1999; 98:675. 30.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1B-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20:6891. 31.Ito K, Jazrawi E, Cosio B, Barnes PJ, Adcock IM. p65 activeted histone acetyltransferase activity is repressed by glucocorticoids: mifepristone fails to recruit HDAC2 to the p65-HAT complex. J Biol Chem 2001; 276:30208. 32.Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 2000; 20:1899. 33.Fryer CJ, Archer TK. Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature 1998; 393:88. 34.Gronemeyer H. Control of transcription activation by steroid hormone receptors. Faseb J 1992; 6:2524. 35.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell 1995; 83:851. 36.Barnes JB, Rodger IW, Thomson NC. Asthma - Basic Mechanisms and Clinical Management . London, Academic Press, 3rd ed., 1998. 37.Ponta H, Cato ACB, Herrlick P. Interference of specific transcription factors. Biochim Biophys Acta 1992; 1129:255. 38.Horvath G, Wanner A. Tracheobronchial Circulation. In: Barnes P, Drazen J, Rennard S, Thomson N. Asthma and COPD Basic Mechanisms and Clinical Management . London: Academic Press; 2002:177-182. 39.Nauck M, Roth M, Tamm M et al. Induction of vascular endothelial growth factor by platelet-activating factor and platelet-derived growth factor is downregulated by corticosteroids. Am J Respir Cell Mol Biol 1997; 16:398. 40.Adock IM, Brown CR, Gelder CM, Shirasaki H, Peters MJ, Barnes PJ. The effects of glucocorticoids on transcription factor activation in human peripheral blood mononuclear cells. Am J Physiol 1995; 37:C331. 41.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF- 6 B and the glucocorticoid receptor. Proc Natl Sci USA 1994; 91:752. 42.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF 6 B and the glucocorticoid receptor. Proc Natl Acad Sci USA 1994; 91:752. 43.Adock IM, Shirasaki H, Gelder CM, et al. The effects of glucocorticoids on phorbol ester and cytokine stimulated transcription factor activation in human lung. Life Sci 1994; 55:1147. 44.Mukaida N, Morita M, Ishikawa Y et al. Novel mechanism of glucocorticoid-mediated gene repression: nuclear factor- 6 B is target for glucocorticoid-mediated interleukin 8 gene repression. J Biol Chem 1994; 269:13289. 45.O'Byrne P. Corticosteroids. In Albert RK, Spiro SG, Jett JR. Comprehensive Respiratory Medicine .London:Mosby,1999. 46.Ristimaki A, Narko K, Hla T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J 1996; 318:325. 47.Bickel M, Iwai Y, Pluznik DH, Cohen RB. Binding of sequence-specific proteins to the adenosine- plus uridine-rich sequences of the murine granulocyte/macrophage colony-stimulating factor mRNA. Proc Natl Acad Sci USA 1992; 89:10001. 48.Adkins KK, Levan TD, Miesfeld RL, Bloom JW. Glucocorticoid regulation of GM-CSF; evidence for transcriptional mechanisms in airway epithelial cells. Am J Physiol 1998; 275: L372. 42.Newton R, Seybold J, Kuitert LM, Bergmann M, Barnes PJ. Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J Biol Chem 1998; 273:323. 50.Wang J, Zhu Z, Nolfo R, Elias JA. Dexamethasone regulation of lung epithelial cell and fibroblast interleukin-11 production. Am J Physiol 1999; 276:L175. 51.Bergmann M, Barnes PJ, Newton R. Molecular regulation of granulocyte macrophage colony-stimulating factor in human lung epithelial cells by interleukin (IL)-1B, IL-4, and IL-13 involves both transcriptional and post-transcriptional mechanisms. Am J Cell Mol Biol 2000; 22:582. 52.Caelles C, Gonzalez-Sancho JM, Monoz A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev 1997; 11:3351. 53.Vanden Berghe W, Vermeulen L, De Wilde G, De Bosscher K, Boone E, Haegeman G. Signa trasnduction by tumor necrosis factor and gene regulation of the inflammatory cytokine interleukin-6. Biochem Pharmacol 2000; 60:1185. 54.Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol 2001; 21:771. 55.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein-kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol 2002; 22:7802. 56.Lewis GD, Campbell WB, Johanson AR. Inhibition of prostaglandin synthesis by glucocorticoids in human endothelial cells. Endocrinology 1986; 119:62. 57.Chung KF, Wiggins J, Collins J. Corticosteroids. In: Bronchial asthma: mechanisms and therapeutics. Boston, Little Brown and Company, 3rd ed., 1993. 58.Abbinante-Nissen JM, Simpson LG, Leikauf GD. Corticosteroids increases secretory leukocyte protease inhibitor transcript levels in airway epithelial cells. Am J Physiol 1995; 12:L601. 59.Borson DB, Gruenert DC. Glucocorticoids induce neutral endopeptidase in transformed human tracheal epithelial cells. Am J Physiol 1991; 260:L83. 60.Wang P, Wu P, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor kappa B activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem 1995; 270:9558. 61.Chernoff AE, Granowitz EV, Shapiro L et al. A randomized, controlled trial of IL-10 in humans. Inhibition of inflammatory cytokine production and immune responses. J Immunol 1995; 154:5492. 62.Naseer T, Minshall EM, Leung DY, et al. Expression of IL-12 and IL-13 mRNA in asthma and their modulation in response to steroid therapy. Am J Respir Crit Care Med 199; 155:845. 63.Levine SJ, Benfield T, Shelhamer JH. Corticosteroids induce intracellular interleukin-1 receptor antagonist type I expression by a human airway epithelial cell line. Am J Respir Cell Mol Biol 1996; 15: 245. 64.Sousa AR, Lane SJ, Nakhosteen JA, Lee TH, Poston RN. Expression of interleukin-1 beta (IL-1 b ) and interleukin-1 receptor antagonist (IL-1ra) on asthmatic bronchial epithelium. Am J Respir Crit Care Med 1996; 154: 1061. 65.Colotta F, Re F, Muzio M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science (Washington DC) 1993; 261: 4725. 66.Mak JCW, Nishikawa M, Barnes PJ. Glucocorticosteroids increase $ 2 -adrenergic receptor transcription in human lung. Am J Physiol 1995; 12:L41. 67.Adocck IM. Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther 2001; 14:211. 68.Ito K, Barnes PJ, Adock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits IL-1B-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20:6891. 69.Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem 2001; 276:29603. 70.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phsophatase-mediated inhibition of MAPK p38. Mol Cell Biol 2002; 22:7802. 71.Gillis S, Crabtree GR, Smith KA. Glucocorticoids-induced inhibition of T-cell growth factor production: I. The effect on mitogen-induced lymphocyte proliferation. J Immunol 1979; 123:1624. 72.Lamas AM, Leon OG, Schleimer RP. Glucocorticoids inhibit eosinophil responses to granulocyte macrophage colony-stimulating factor. J Immunol 1994; 147:254. 73.Wallen IL, et al. Glucocorticoids inhibit cytokine-mediated eosinophil survival. J Immunol 1991; 147:3490. 74.Altman LC, et al. Effect of corticosteroids on eosinophil chemotaxis and adherence. J Clin Invest 1981; 67:28. 75.Colotta F, Re F, Muzio M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulate by IL-4. Science 1993; 261:472. 76.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leucocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA 1992; 89:9991. 77.Weiss EB, Stein M. Bronchial Asthma. Little Brown and Company, 3rd ed., Boston, 1993. 78.Adock IM, Peters M, Gelder C, Shirasaki H, Brown CR, Barnes PJ. Increased tachykinin receptor gene expression in asthmatic lung and its modulation by steroids. J Mol Endocrinol 1993; 11:1. 79.Knowles RG, Salter M, Brooks SL, Moncada S. Anti-inflammatory glucocorticosteroids inhibit the induction by endotoxin of nitric oxide synthase in the lung, liver and aorta of the rat. Biochem Biophys Res Commun 1990; 172:1042. 80.Nelson BV, Sears S, Woods J et al. Expired nitric oxide as a marker for childhood asthma. J Pediatr 1997; 130:423. 81.Baraldi E, Azzolin NM, Zanconato S, Dario C, Zacchello F. Corticosteroids decrease exhaled nitric oxide in children with acute asthma. J Pediatr 1997; 131:381. 82.Mitchell JA, Belvisi MG, Akarasereemom P, et al. Induction of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. Br J Pharmacol 1994; 113: 1008. 83.Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kB and nuclear factor-interleukin 6 in the tumor necrosis- a -dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem 1995; 270: 31315. 84 Newton R, Kuitert LM, Slater DM, Adcock IM, Barnes PJ. Induction of cPLA 2 and COX-2 mRNA by proinflammatory cytokines is suppressed by dexamethasone in human airway epithelial cells. Life Sci 199784.Reisman D, Thompson EA. Glucocorticoid regulation of cyclin D3 gene transcription and mRNA stability in lymphoid cells. Mol Endocrinol 1995; 9:1500. ; 60: 67. 85.Yanagisawa M, Kurihara H, Kimura, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332:411. 86.Vittori E, Marini M, Fasoli A. Increased expression of endothelin in bronchial epithelial cells of asthmatic patients and effect of corticosteroids. Am Rev Respir Dis 1992; 146:1320. 87.Reisman D, Thompson EA. Glucocorticoid regulation of cyclin D3 gene transcription and mRNA stability in lymphoid cells. Mol Endocrinol 1995; 9:1500. Última Atualização: - 16/03/2019 |