A contratilidade muscular é definida como a relação instantânea entre força, velocidade, comprimento e tempo.1 A contratilidade exacerbada das células musculares lisas das vias aéreas é uma característica da asma e foi bem demonstrada por Ma et al. através de células isoladas por meio de biópsias endobrônquicas. Aumentos estatisticamente significativos na capacidade máxima de encurtamento foram encontrados em células de músculo liso brônquico de indivíduos com asma em comparação às células normais.2
Na asma ocorrem alterações nas propriedades contráteis do músculo liso das vias aéreas simultaneamente ao aumento em sua massa devido à hipertrofia e/ou hiperplasia, que são presumivelmente os principais responsáveis pelo aumento da responsividade das vias aéreas. Isso as leva a se contraírem mais facilmente em resposta a estímulos, podendo se observar a presença de tônus persistente aumentado da musculatura lisa das vias aéreas nestes pacientes.3-5 É provável que essas alterações ocorram precocemente na história natural da asma.
A contração do músculo liso brônquico decorre da interação de agonistas contráteis como a acetilcolina (Ach), histamina, endotelina-1 (ET-1), prostaglandinas e leucotrienos que determinam a broncoconstrição através de receptores acoplados à proteína G (GPCR). Nas células musculares lisas, a transdução de sinal ocorre quando esses mediadores se ligam a seus receptores específicos na membrana. (Figura 1)
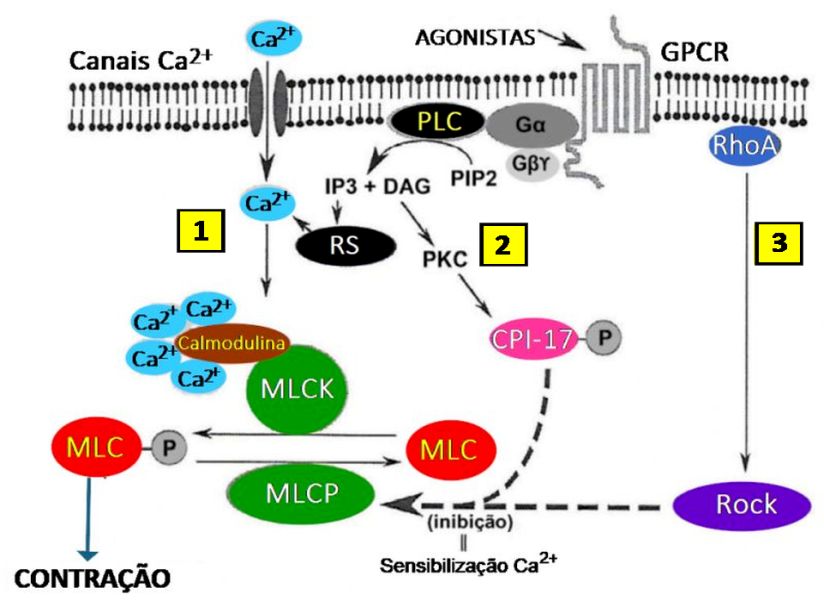
Figura 1 – Regulação da Contração do Músculo Liso
DAG – diacilglicerol; GPCR – receptor acoplado à proteína G; IP3 – trifosfato de inositol; MLC – cadeia leve de miosina; MLCK – quinase de cadeia leve de miosina; MLCP – fosfatase de cadeia leve de miosina; PKC – proteína quinase C; PLC – fosfolipase C; PIP2 – fosfatidilinositol-4,5-bifosfato; ROCK – RHO-associated, Coiled-coil Containing Protein Kinase ou Rho-kinase (Rho quinase; RhoA, família Rho pequena GTPase A); RS – retículo sarcoplasmático. Figura retirada e modificada de Chiba Y et al. J Pharmacol Sci. 2010; 114:239-47.
Quando os GPCRs são ativados por agonistas broncoconstritores, ocorre o acoplamento com uma proteína G heterodimérica (composta por subunidades a, β e γ), promovendo a troca de Difosfato de Guanosina (GDP) por Trifosfato de Guanosina (GTP) na subunidade a. Isso induz à dissociação do complexo, liberando a subunidade Gag/11 ativada, que ativa a enzima fosfolipase C-ß (PLC-ß) (principal via canônica da indução de contração).
➭ O evento desencadeante para qualquer contração muscular é um aumento dos íons cálcio (Ca2+) intracelulares. Após a estimulação pelos agonistas contráteis acontece a abertura dos canais de Ca2+, promovendo a entrada de íons Ca2+, que irão promover a ativação da contração. A origem do cálcio é o líquido extracelular e o retículo sarcoplasmático (RS).
1 – A Gag/11 ativa a fosfolipase C-ß, que hidrolisa o PIP2 (fosfatidilinoditol-4,5-bifosfato) em dois segundos mensageiros:
■ IP3 (inositol trifosfato) ⇒ liga-se a receptores IP3R no retículo sarcoplasmático (RS), liberando Ca2+ para o citosol6-9. O Ca2+ se liga à calmodulina (CaM), uma proteína mensageira ligadora de cálcio, e o aumento do Ca2+ forma o complexo homo tetramétrico constituído por quatro subunidades, cada uma com peso molecular de 260 kDa.10
■ DAG (diacilglicerol) ⇒ permanece na membrana e ativa a proteína quinase C (PKC).
O complexo 4Ca2+– calmodulina – miosina quinase de cadeia leve (MLCK) em contrapartida abre e ativa o domínio enzimático da MLCK.8 A atividade da quinase de cadeia leve de miosina (MLCK) é controlada pela elevação na concentração do Ca2+ citosólico proveniente do influxo de cálcio extracelular dos reservatórios do RS.
A capacidade contrátil das células musculares lisas presentes nas vias aéreas é primordialmente influenciada pela fosforilação da cadeia leve de miosina (MLC) – "fosforilar" significa adicionar um grupo fosfato. Essa fosforilação é o evento-chave para iniciar o ciclo da ponte cruzada de actina-miosina e assim a contração. O nível de fosforilação da MLC é meticulosamente controlado por meio da ação de duas enzimas determinantes: a cadeia leve de miosina quinase (MLCK) e a fosfatase de cadeia leve de miosina (MLCP).6
O aumento do Ca2+ intracelular é o evento central que desencadeia a cascata de contração através da via Ca2+/calmodulina ⇒ MLCK ⇒ fosforilação da MLC |
|
A MLCK se liga às moléculas de MLC nas fibras de miosina dentro da célula muscular lisa. Inicia-se a transferência de grupos fosfato de ATP (trifosfato de adenosina) para a MLC. Isso resulta na fosforilação da MLC – resíduo de aminoácido específico (serina 19) da subunidade reguladora da cadeia leve de 20 kDa de miosina – que é uma modificação química importante. A fosforilação promove a formação de pontes cruzadas com o filamento de actina e sofre transformações moleculares cíclicas, permitindo que os filamentos de miosina se liguem aos filamentos de actina encurtando11 a célula muscular lisa, resultando na sua contração.6-8,12 A subunidade ligada à miosina, quando fosforilada, inibe a atividade enzimática da MLCP, permitindo que a MLC se mantenha fosforilada, promovendo assim a contração do músculo liso.6,7,13,14 (Figura 1)
No entanto, duas vias paralelas podem inibir essa desfosforilação:
2 – Após a hidrólise do fosfatidilinositol-4,5-bisfosfato (PIP2) pela PLC o diacilglicerol ativa a proteína quinase C (PKC) que sequencialmente leva à fosforilação da proteína inibidora da fosfatase (MLC) potencializada pela proteína quinase C de 17 kDa (CPI-17), para inibição do MLCP e favorecer a contração6,15 — outra via da sensibilização ao Ca2+. (Figura 1)
3 – Agonistas contráteis atuando em GPCRs acoplados à proteína G levam conjuntamente à elevação de RhoA e Rho-quinase (ROCK), o que também conduz à subsequente inibição da MLCP.6,7,15 Mantém a MLC fosforilada independentemente do Ca2+ – fenômeno de sensibilização do Ca2+ – ou seja, promove contração sustentada sem aumento adicional de Ca2+. A via da RhoA/Rho-quinase, onde a Rho-quinase uma serina/treonina quinase, é ativada por agonistas como endotelina-1 via Ga12/13.
A MLCP é uma serina (Ser)-treonina (Thr) fosfatase expressa no músculo liso cuja função é independente dos níveis de Ca2+. A MLCP é a enzima responsável pela desfosforilação da MLC. Portanto, a ação da MLCP, que remove os grupos fosfato da MLC, ⇒ relaxa o músculo liso. A MLCP é uma enzima complexa composta por várias subunidades. As principais subunidades incluem a catalítica PP1c (Proteína Fosfatase 1 catalítica), as regulatórias Myosin Phosphatase Target Subunit 1(MYPT1) e MYPT2 e outras subunidades acessórias.16 A atividade da MLCP é regulada por meio da fosforilação de sua subunidade MYPT1.
A Rho-quinase atua fosforilando a subunidade regulatória (ou subunidade M) da MLCP, a MYPT1. Em particular, a fosforilação de MYPT1 nos resíduos Ser507, Thr853/850 e Thr696/695 medeia a inibição da atividade de MLCP e, portanto, a inibição do relaxamento do músculo liso das vias aéreas.17 Além disso, a fosforilação do MYPT1 em Ser965, Ser696 e Ser852 também é observada no músculo liso das vias aéreas. Essa fosforilação inibindo a MLCP promove assim o estado fosforilado da MLC8 e, consequentemente, ⇒ a contração do músculo liso – é a sensibilização ao Ca2+. (Figura 1)
A via de sensibilização ao Ca2+ leva à contração máxima sem levar em conta a concentração intracelular de Ca2+, pela regulação do estado de fosforilação da MLC pela MLCP.9
Além da liberação do retículo, ocorre abertura de canais de Ca2+ dependentes de voltagem e canais ROC (operados por receptor).
A Rho-quinase atua fosforilando a subunidade M regulatória da MLCP, a MYPT1, suprimindo a sua atividade, gerando consequente contração do músculo liso. |
|
➭ Como resultado de um processo de homeostase, o aumento nos níveis citosólicos livres de Ca2+ é então rápido e praticamente revertido por uma recaptura no retículo sarcoplasmático/endoplasmático, basicamente mediada pela ATPase de cálcio do retículo sarcoplasmático/endoplasmático (SERCA), que reabastece assim as reservas intracelulares de Ca2+ previamente esgotadas.14 Esse processo de transporte de cálcio pelo SERCA é fundamental para regular a concentração de cálcio no citoplasma, permitindo que as células musculares relaxem após a contração, controlando a liberação de cálcio durante a contração muscular e desempenhando papel importante em muitos outros processos celulares, como a sinalização celular e a homeostase de cálcio. A energia fornecida pela hidrólise do ATP é essencial para impulsionar esse processo contra um gradiente de concentração.
A remoção do Ca2+ do citosol através de processo energético dependente de ATP, mediado pela bomba SERCA (recaptura) viabiliza o relaxamento e o término da contração. Modificações na função ou expressão SERCA estão envolvidas na fisiopatologia da asma em relação ao tônus do músculo liso peribrônquico, contribuindo para o relaxamento dificultado e agravando a hiper-responsividade das vias aéreas (HVA).
O término da contração e o relaxamento do músculo liso são possibilitados principalmente pela remoção do Ca2+ citosólico, um processo energético (dependente de ATP) mediado pela bomba SERCA no retículo sarcoplasmático. Alterações na função ou expressão do SERCA estão implicadas na fisiopatologia da asma, contribuindo para o relaxamento prejudicado e a HRB. Na asma ocorre uma expressão diminuída do SERCA no músculo liso das vias aéreas com importantes repercussões:18,19
1) Uma seria o relaxamento prejudicado e lentificado, traduzido por menos bombas SERCA funcionando, gerando remoção de Ca2+ do citosol mais lenta e ineficiente, que ocasionam contrações mais prolongadas além de dificuldade para o músculo relaxar plenamente após estímulos constritores.20 Por outro lado, o músculo sensibilizado, responde de forma exagerada a novos estímulos (HVA), devido ao fato do Ca2+ basal encontrar-se mais elevado.
2) A outra consequência seria relacionada à recuperação dos estoques de Ca2+ do RS após a contração, pois o SERCA é determinante para o seu reabastecimento. Na asma a redução do SERCA impede o reabastecimento do RS e a célula interpreta esse "RS vazio" como um sinal para abrir canais de Ca2+ na membrana o que contraditoriamente pode estimular mecanismos compensatórios como um sinal para abrir canais de Ca2+ na membrana resultando em aumento sustentado de Ca2+ citosólico, hiperexcitabilidade e broncoconstrição.
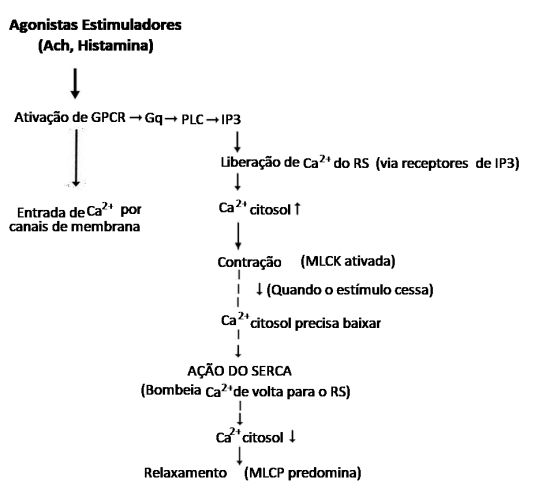
Figura 2 – Fluxograma Integrado – Contração X Relaxamento