|
Asma
INFECÇÃO VIRAL RESPIRATÓRIA E ASMA
As infecções do trato respiratório superior constituem-se nas infecções mais comuns dos seres humanos1 e representam a causa mais frequente de consulta médica aos clínicos.
As infecções virais têm sido implicadas como um importante fator tanto no desenvolvimento da asma como na indução de suas exacerbações.2 A associação entre infecção viral e episódios agudos de asma foi pela primeira vez mencionada durante a pandemia de gripe de 1957.3,4
Crianças em idade pré-escolar apresentam 6 a 10 resfriados por ano, enquanto os adultos apresentam em média três episódios,5 presumivelmente devido ao desenvolvimento de imunidade contra os principais microrganismos associados às infecções. As infecções virais aumentam o absenteísmo ao trabalho e à escola, apresentando efeitos adversos em pacientes com doença broncopulmonar subjacente. Dados epidemiológicos dos EUA estimam em no mínimo 20 milhões as faltas ao trabalho e em 22 milhões o absenteísmo escolar a cada ano, em consequência de infecções virais respiratórias.6,7 No início da década de 2000 Fendrick et al. estimaram em US$ 40 bilhões por ano o impacto econômico total da infecção viral do trato respiratório alto, não relacionado à gripe nos Estados Unidos por ano.8
As infecções virais respiratórias altas e a atopia interagem de forma bidimensional e dinâmica. As infecções virais influenciam o desenvolvimento de sensibilização alergênica, enquanto que a atopia influencia as respostas das vias aéreas respiratórias baixas à infecção viral.9 Os pacientes com asma são mais susceptíveis a infecções virais respiratórias do que os não asmáticos, apresentando uma resposta nitidamente mais forte.10 Estudos epidemiológicos prospectivos evidenciam que 80% das exacerbações de asma em crianças em idade escolar e 50% de todas as exacerbações em adultos estavam associadas a infecções virais do trato respiratório superior, sendo o rinovírus humano considerado como o principal vírus associado a exacerbações em asmáticos.11 Existem poucas dúvidas de que a maioria dos vírus (adenovírus, influenza, vírus sincicial respiratório, parainfluenza, coxsackievírus) infecta o trato respiratório baixo.12 Existem evidências muito claras de que isto ocorra também com os rinovírus. Pattemore et al.13 detectaram vírus em secreções brônquicas em mais de 40% das exacerbações de asma em crianças e em mais de 20% das exacerbações em adultos. Esta mesma avaliação durante a fase assintomática em asmáticos e não asmáticos não ultrapassou os 3%.14-16
Alguns vírus, como o vírus sincicial respiratório (VSR), o parainfluenza (PIV), influenza, coronavírus e o rinovírus (RV), parecem ter um papel mais importante que outros vírus na exacerbação da asma, podendo atuar também como alérgenos, estimulando a formação de anticorpos IgE vírus-específicos. Além disso, infecções virais podem potencializar a resposta subsequente à inalação alergênica. Existem fortes evidências que demonstram que a infecção pelo RV aumenta a responsividade brônquica em alérgicos e naqueles com asma. Estas mudanças na responsividade brônquica se iniciam 2 a 3 dias após a exposição ao vírus e persistem por vários dias e às vezes por semanas após a infecção.17,18 Em 10–20% dos pacientes é possível obter o vírus em cultura de secreções 2 a 3 semanas após o início da infecção.6 Arruda et al.19 relataram um tempo médio de infecção pelo RV entre 9,5 e 11 dias. Em certas faixas etárias, como nos idosos, este tempo pode ser mais longo, alcançando 16 dias.20
Estudos utilizando RT–PCR (Reverse Transcription–Polymerase Chain Reaction) detectaram vírus em mais de 80% das crises de asma em crianças entre 9 e 11 anos, acompanhadas por um período de dois anos, estando o RV (família Picornaviridae – genoma RNA) associado à quase 60% das exacerbações.21 Em outro estudo, desta feita com adultos, também por 2 anos, Nicholson et al.22 detectaram sintomas de 'resfriados' em 71% das exacerbações da doença e redução nos valores do pico de fluxo expiratório (PFE) em 27%. Nos casos em que o agente etiológico foi identificado, o RV foi o responsável por 57% dos casos de agudização da asma, e destes, 23% apresentaram reduções no PFE ≥ 50 l/min.22
Em crianças a infecção viral comumente causa sibilos. Mais de 70% dos episódios de sibilância no primeiro ano de vida estão relacionados a infecções virais respiratórias.23 Nas menores de 2 anos o agente causal mais comum é o ubíquo vírus sincicial respiratório (família Paramyxoviridae – genoma RNA), sendo também o maior agente causador das bronquiolites (80%) (Figura 1). Os vírus circulantes foram classificados em dois grupos antigênicos (A e B), sendo que o VSR-A está associado à maior morbidade.24 Muitas vezes torna-se difícil identificar se a criança apresenta crise aguda de exacerbação de asma ou bronquiolite. O VSR é extremamente contagioso e a transmissão ocorre por contato direto com secreções contaminadas ou fômites (objetos contaminados por vírus).
 O mecanismo pelo qual o VSR desencadeia a sibilância está associado a uma resposta de células T caracterizada principalmente pela produção de citocinas Th2, a mesma resposta observada durante os episódios de asma. Ambas são caracterizadas pelo recrutamento de células T e eosinófilos bem como pela liberação de mediadores solúveis como a histamina, as cininas e os leucotrienos. Entre as crianças com bronquiolite, a sibilância mais severa e frequente correlaciona-se com os elevados níveis de anticorpos IgE contra o VSR e o vírus parainfluenza nas secreções respiratórias, sugerindo que os anticorpos induzidos pelos vírus aumentam a liberação de mediadores inflamatórios, importantes na responsividade brônquica. O VSR pode favorecer ainda o broncospasmo através de vias neurais que medeiam a responsividade das vias aéreas. O mecanismo pelo qual o VSR desencadeia a sibilância está associado a uma resposta de células T caracterizada principalmente pela produção de citocinas Th2, a mesma resposta observada durante os episódios de asma. Ambas são caracterizadas pelo recrutamento de células T e eosinófilos bem como pela liberação de mediadores solúveis como a histamina, as cininas e os leucotrienos. Entre as crianças com bronquiolite, a sibilância mais severa e frequente correlaciona-se com os elevados níveis de anticorpos IgE contra o VSR e o vírus parainfluenza nas secreções respiratórias, sugerindo que os anticorpos induzidos pelos vírus aumentam a liberação de mediadores inflamatórios, importantes na responsividade brônquica. O VSR pode favorecer ainda o broncospasmo através de vias neurais que medeiam a responsividade das vias aéreas.
Avaliações apontam que a doença por VSR resulta em mais de 30 milhões de casos de infecção do trato respiratório baixo em crianças menores de cinco anos por ano em todo o mundo a cada ano, com 3,2 milhões de hospitalizações e 200.000 mortes anuais em todo o mundo.25 Nos EUA a bronquiolite é a principal causa de internações em lactentes, alcançando 110.000 hospitalizações anuais.26 Aproximadamente uma em cada cinco crianças sibilam em decorrência da infecção pelo VSR. Há longa data a bronquiolite é reconhecida como um fator de risco para o desenvolvimento de asma infantil persistente. Cerca de 30% dos lactentes hospitalizados com bronquiolite grave desenvolverão posteriormente asma infantil.27-29 O porquê ainda não está bem estabelecido, embora existam hipóteses. Acredita-se que a bronquiolite inflamatória pós-infecção tenha forte potencial de injúria ao nível das vias aéreas, determinando o seu remodelamento. Estas alterações permanentes influenciariam o crescimento "pulmonar", causando sibilância persistente. Outra possibilidade propõe que a infecção pelo VSR, em indivíduos geneticamente propensos à atopia, possa influenciar o desenvolvimento do sistema imune, tornando o paciente mais alérgico.
Fujiogi et al.30 analisando três estudos de coorte prospectivos multicêntricos que incluíram 3,081 lactentes hospitalizados (idade <12 meses) por bronquiolite grave identificaram quatro perfis clinicamente distintos e reprodutíveis de bebês hospitalizados pela doença. O objetivo era determinar qual grupo teria maior probabilidade de desenvolver asma infantil tardiamente, ao longo dos anos. Concluíram pelo perfil que incluía: crianças com histórico de problemas respiratórios prévios e eczema, infecção por rinovírus e baixa prevalência para infecção pelo VSR. Este grupo representou 13% das coortes e apresentou a maior probabilidade de desenvolver asma com 2,5 vezes mais chances em comparação com o perfil clássico de bronquiolite". Na análise geral, o risco de desenvolver asma aos 6 ou 7 anos foi de 23%.30
Um estudo prospectivo sobre a associação entre receptores Toll-like (TLR) e o surgimento de asma infantil tardia após bronquiolite desenvolvido por Korppi & Törmänen na Finlândia, concluiu que as variações genéticas em receptores TLR1 e TLR10 foram associadas a mais asma em crianças com 5–7 anos e 11–13 anos, com resultados inconsistentes para os outros oito genes TLRs.31
Embora o VSR acometa principalmente as crianças menores de 2 anos, deve ser salientado que infecções por este vírus podem configurar sério problema no outro extremo, em pacientes idosos.
Acima de 2 anos até a idade adulta, as infecções virais são causadas principalmente pelos rinovírus (Figura 2) e coronavírus (no hemisfério norte é mais prevalente no outono e inverno), ocorrendo forte associação entre RV e asma.21,32 O Brasil não dispõe de dados epidemiológicos regulares para estabelecer padrões sazonais desses vírus.
A família dos Picornaviridae é a fonte mais comum de infecções virais no mundo.33 Recebem este nome devido ao seu ínfimo tamanho, consistindo em aproximadamente 7.200 pares de bases. São vírus RNA, ubíquos e incluem além dos rinovírus humanos, os enterovírus, cardiovírus e os aphtovírus.34 Os rinovírus compartilham propriedades básicas com os enterovírus, incluindo 40–60% de homologia entre seus genomas.35 O RV é um vírus sem envelope, com forma icosaédrica (poliedro com 20 lados) com diâmetro 20–27nm. É o responsável por cerca de 60% dos resfriados comuns.
Mais de 160 tipos de rinovírus geneticamente distintos pertencentes a três espécies (A, B e C) são agora reconhecidos. O rinovírus se replica principalmente nas células epiteliais que revestem o trato respiratório superior e inferior.
 São classificados em três espécies genéticas baseados na sequência homológica. Oitenta sorotipos/genótipos são classificados como RV-A, enquanto o RV-B compreende trinta e dois membros. Os sorotipos restantes pertencem à mais recente descoberta RV-C que compreende cinquenta e sete sorotipos.36,37 As exacerbações da asma nas crianças menores de dez anos de idade e hospitalizadas estão mais relacionadas ao RV-C. O RV-C está sempre associado à internação de crianças com asma moderada/severa.38 Em vários estudos a exacerbação pelo RV-B foi menos frequente e menos severa. O RV infecta pacientes de qualquer idade, em qualquer época do ano, sendo mais prevalente, entretanto, no inverno. Adenovírus, enterovírus e coronavírus são detectados com menos frequência. São classificados em três espécies genéticas baseados na sequência homológica. Oitenta sorotipos/genótipos são classificados como RV-A, enquanto o RV-B compreende trinta e dois membros. Os sorotipos restantes pertencem à mais recente descoberta RV-C que compreende cinquenta e sete sorotipos.36,37 As exacerbações da asma nas crianças menores de dez anos de idade e hospitalizadas estão mais relacionadas ao RV-C. O RV-C está sempre associado à internação de crianças com asma moderada/severa.38 Em vários estudos a exacerbação pelo RV-B foi menos frequente e menos severa. O RV infecta pacientes de qualquer idade, em qualquer época do ano, sendo mais prevalente, entretanto, no inverno. Adenovírus, enterovírus e coronavírus são detectados com menos frequência.
Os principais sorotipos de RV-A e RV-B entram predominantemente no epitélio ao longo do trato respiratório através da molécula de adesão intercelular 1 (ICAM-1) e representam a via de entrada para mais de 90% de todas as infecções por RV.39
A indução viral deficiente e tardia de IFN-α, IFN-ß, IFN-Υ e IFN-λ em células dendríticas plasmocitoides (pDCs), células do lavado broncoalveolar (BAL) e células epiteliais brônquicas primárias foi bem caracterizada na asma,40-43 o que pode resultar em maior carga viral e em 'atraso' na eliminação do RV, com maior carga viral, contribuindo para exacerbações, doenças potencialmente mais graves43 com uma superexpressão de um ambiente inflamatório Th2.44
O coronavírus causa sintomas semelhantes ao rinovírus, sendo responsável por aproximadamente 15–20% dos resfriados comuns, determinando mais infecções do trato respiratório inferior do que superior, estando associado frequentemente a exacerbações da asma. Existem dois grupos antigênicos principais de coronavírus, conhecidos como 229E e OC43. O Coronavírus-19 é discutido em capítulo à parte.
 Os vírus influenza pertencem à família Orthomyxoviridae – genoma RNA que contém dois gêneros: influenza vírus Tipo A e influenza vírus Tipo B. O influenza vírus Tipo C representa um outro sem relevância clínica para humanos e geralmente leva a infecções brandas.45 O vírus A influenza (IAV) apresenta dois grupos antigênicos de peplômeros com glicoproteínas em sua superfície externa (H e N) (Figura 3). O grupo da hemaglutinina é caracterizado por proteínas de superfície que atuam como receptores do vírus e induzem a respostas imunes. E o da neuraminidase, uma enzima na superfície das partículas do vírus que destrói o ácido neuramínico (siálico), que faz parte do receptor do vírus para se ligar às células e induz a respostas imunes nos hospedeiros infectados. É a caracterização do tipo de hemaglutininas (H1–H16) e da neuraminidase (N1–N9) que estabelece quais subtipos estão circulando na população mundial (p. ex. H1N1; H3N2). Os vírus influenza pertencem à família Orthomyxoviridae – genoma RNA que contém dois gêneros: influenza vírus Tipo A e influenza vírus Tipo B. O influenza vírus Tipo C representa um outro sem relevância clínica para humanos e geralmente leva a infecções brandas.45 O vírus A influenza (IAV) apresenta dois grupos antigênicos de peplômeros com glicoproteínas em sua superfície externa (H e N) (Figura 3). O grupo da hemaglutinina é caracterizado por proteínas de superfície que atuam como receptores do vírus e induzem a respostas imunes. E o da neuraminidase, uma enzima na superfície das partículas do vírus que destrói o ácido neuramínico (siálico), que faz parte do receptor do vírus para se ligar às células e induz a respostas imunes nos hospedeiros infectados. É a caracterização do tipo de hemaglutininas (H1–H16) e da neuraminidase (N1–N9) que estabelece quais subtipos estão circulando na população mundial (p. ex. H1N1; H3N2).
Uma das características mais importantes dos vírus influenza é a frequência com a qual mudam sua antigenicidade (variação antigênica). São responsáveis por sintomas mais prolongados e severos e apresentam maior mortalidade quando comparada à dos vírus do resfriado comum, pois podem determinar um espectro de doença que vai desde uma simples gripe, ao agravamento da asma até uma doença sistêmica. A pneumonia é a complicação grave mais frequente da gripe. Dois padrões clínicos principais são descritos: pneumonia viral primária e pneumonia bacteriana secundária. A variação antigênica é muito frequente com o vírus A, quase que anual, ocorrendo menos vezes com o vírus Tipo B. O fenômeno das alterações antigênicas (antigenic drifts) resultantes de mutações pontuais durante a replicação viral, ajuda a explicar por que a influenza continua a ser a maior doença epidêmica a acometer o homem, implicando modificações na composição da vacina a cada ano.46 O vírus influenza como causador da exacerbação da asma é o menos frequente, ocorrendo somente durante a epidemia anual.
Mycoplasma pneumoniae e Chlamydia pneumoniae estão também entre os agentes isolados tanto nos resfriados como nas exacerbações da asma, embora a relativa proporção varie consideravelmente entre os estudos.
Nos últimos anos, novos vírus respiratórios foram identificados como agentes potenciais desencadeantes das exacerbações da asma. O metapneumovírus humano (hMPV) foi detectado em mais de 7% de adultos hospitalizados por agudização da asma.47 O hMPV, um Paramyxoviridae – genoma RNA, foi descoberto em 2001 e classificado como do gênero Metapneumovírus. Seu perfil epidemiológico e suas manifestações clínicas são semelhantes às do VSR, tendo sido detectado em 16% de crianças hospitalizadas por bronquiolite.48 Outro vírus, identificado em 1997, é o vírus DNA Torqueteno (TTV), classificado em um novo gênero, Anellovírus. Estudos detectaram a sua presença em secreção nasal de crianças com asma de leve a moderada, havendo correlação entre o TTV nasal e a resistência das vias aéreas.49
Os vírus respiratórios são não móveis, porém podem ser transmitidos via partículas quando a pessoa infectada espirra, assoa ou tosse. A infecção ocorre quando estas partículas são inaladas por outra pessoa e se depositam nas mucosas nasal, faringiana ou do trato respiratório inferior. Partículas menores de 5–10 µm de diâmetro se depositam preferencialmente no trato respiratório baixo, enquanto partículas ≥10–20 µm de diâmetro depositam-se na traqueia ou no trato respiratório superior.
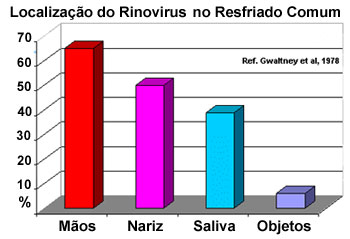
O contágio viral pode ocorrer por contato pessoa a pessoa através do toque interpessoal das superfícies das mãos que tiveram contato com secreções nasais infectadas, para os olhos, nariz ou boca — seguido de autoinoculação, um ciclo que pode ser interrompido pela lavagem da mãos, sendo que a maioria dos vírus respiratórios permanece viável nas superfícies por várias horas, incluindo fômites (Figura 4). Acredita-se que o RV se espalhe no epitélio respiratório por extensão local. Em resfriados experimentais, a nasofaringe posterior é o local de infecção mais intensa.
Novas pesquisas, estimuladas pela pandemia de COVID-19, sugerem que a maioria dos vírus respiratórios também é transmitida pelo ar (microgotículas chamadas aerossóis). Os sintomas ocorrem 16 horas após a inoculação, com um pico 24 a 48 horas.
O epitélio brônquico não atua simplesmente como uma barreira física, apresentando importante papel regulador. As células epiteliais contribuem para a resposta imune que se segue à infecção viral. A extensão do dano epitelial observado no epitélio varia de acordo com o tipo do vírus. O influenza causa tipicamente uma extensa necrose, enquanto que o RV causa apenas pequeno dano, sem ações citotóxicas. A destruição das células epiteliais resulta em aumento da permeabilidade, o que favorece a penetração de agentes irritantes e alérgenos, além de expor uma extensa rede de fibras nervosas aferentes. Estes fatores contribuem para um aumento na hiper-responsividade brônquica. Acredita-se que as células epiteliais possam agir como células apresentadoras de antígenos (APCs), principalmente durante infecções virais respiratórias secundárias. Além disso, as células epiteliais expressam o complexo maior de histocompatibilidade classe I e moléculas coestimulatórias B-7-1 e B-7-2, sendo esta expressão upregulated in vitro pelo RV-16.
Uma orquestração eficiente da resposta imune pelas citocinas é essencial para a erradicação dos vírus. Modificações na expressão de citocinas das vias aéreas pode contribuir para aumento na gravidade da infecção viral na asma. Quando os vírus penetram nas células epiteliais, eles iniciam uma série de alterações, tornando mais intensa a responsividade brônquica. Estudos in vitro evidenciaram que as células epiteliais quando infectadas pelo RV são induzidas a produzir citocinas como a IL-1a, IL-6, IL-8, IL-11, MIP-1a e GM-CSF.50-52 Pesquisas demonstraram que as células do epitélio brônquico, quando infectadas pelo RV, são induzidas também a produzir citocinas quimiotáxicas como a eotaxina e RANTES que desempenham função de recrutamento e ativação dos eosinófilos nas vias aéreas. RANTES é quimiotáxico para eosinófilos, linfócitos T, monócitos e basófilos.53,54 A eotaxina é seletiva para os eosinófilos, não tendo ação sobre os linfócitos, monócitos ou neutrófilos.55,56 Os neutrófilos são precocemente recrutados na infecção viral em resposta à produção de IL-8 pelas células epiteliais e neutrófilos ativados, sendo sua presença proeminente na asma severa. Estudos com escarro induzido em pacientes com asma e voluntários não asmáticos demonstram um aumento significante de neutrófilos no quarto dia de resfriado comum, correlacionando-se com a IL-8 do escarro. O ENAP-78 (epiThelial neutrophil-activating peptide-78) que induz à migração de neutrófilos encontra-se com concentrações elevadas em amostras de secreção nasal de pacientes infectados com RV.57 A expressão ICAM-1 está aumentada nas células do epitélio brônquico em pacientes com asma (via mecanismo que envolve o NF-kB), quando comparada a normais e a pacientes com bronquite crônica.58 Este aumento na expressão ICAM-1 parece ampliar a susceptibilidade destas células à infecção pelo RV.59 Outrossim, infecções virais, incluindo o parainfluenza, aumentam a expressão endotelial do VCAM-1, que interage com a ß1 integrina VLA-4, importante molécula de adesão na migração de linfócitos e eosinófilos.
Dados têm demonstrado que a infecção pelo RV pode estimular a produção pelo epitélio de fatores angiogênicos e fibroproliferativos, como o VEGF (vascular endoThelial growTh factor) e FGF-2 (fibroblast growTh factor),60 que contribuem para mudanças estruturais brônquicas relacionadas ao remodelamento da asma.
Nas células epiteliais normais, a infecção viral induz à produção de interferon beta (IFN-ß), que induz à apoptose das  células infectadas pelos vírus e limita sua replicação. O IFN-ß é uma citocina antiviral (Tipo I) produzida por células infectadas pelo RV humano que apresenta efeitos antivirais diretos, limitando a replicação viral ao induzir um estado antiviral em células infectadas e em células vizinhas através da ativação de genes estimulados por IFN (Interferon-Stimulated Genes – ISGs).61 Respostas deficientes de interferon foram preditivas da gravidade das exacerbações induzidas por vírus, sendo que estudos demonstram que asmáticos expressam cargas virais mais elevadas durante infecções por RV.61-63 A atuação principal do IFN-ß na inflamação viral verifica-se em duas frentes: células infectadas pelos vírus e limita sua replicação. O IFN-ß é uma citocina antiviral (Tipo I) produzida por células infectadas pelo RV humano que apresenta efeitos antivirais diretos, limitando a replicação viral ao induzir um estado antiviral em células infectadas e em células vizinhas através da ativação de genes estimulados por IFN (Interferon-Stimulated Genes – ISGs).61 Respostas deficientes de interferon foram preditivas da gravidade das exacerbações induzidas por vírus, sendo que estudos demonstram que asmáticos expressam cargas virais mais elevadas durante infecções por RV.61-63 A atuação principal do IFN-ß na inflamação viral verifica-se em duas frentes:
1. ■ Através de efeito antiviral direto pela via de sinalização JAK/STAT – O IFN-ß exerce sua atuação através de uma cascata clássica de fosforilação que converte o sinal extracelular em resposta transcricional.64,65
|
o IFN-ß se liga ao receptor heterodímero IFNAR1/IFNAR2. |
|
a ligação ativa as Janus quinases associadas: JAK1 e TYK2. |
|
essas quinases fosforilam as proteínas STAT1 e STAT2, que se dimerizam e recrutam o IRF9 (Fator Regulador de Interferon 9). Juntos eles formam o complexo ISGF3. |
● |
o complexo ISGF3 migra para o núcleo e se liga aos Elementos de Resposta Estimulados por Interferon (ISRE) nos promotores de Genes Estimulados por Interferon (ISGS). Esses genes bloqueiam a replicação viral, p.ex., degradando RNA viral (via OAS/RNAase L) ou inibindo a tradução (via PKR). |
2. ■ Modulação da inflamação – O IFN-ß reduz a liberação excessiva de citocinas inflamatórias (como IL-6, TNF-α e quimiocinas CXCL10/CCL5) que recrutam neutrófilos e linfócitos. Com isso, ele limita o dano tecidual e os sintomas de obstrução nasal e exacerbação de doenças como a asma. Diferente de um anti-inflamatório direto, o IFN-ß atua principalmente bloqueando a causa determinante da inflamação, a replicação viral, e modulando a cinética da resposta imune inata e adaptativa. A supressão desta replicação é alcançada através de ISGs como OAS (2'-5'-oligoadenilato sintetase) e MX1 que degradam o RNA viral e bloqueiam a montagem do vírus. Havendo menos vírus ocorre menos estímulo para indução e liberação de citocinas 66
Entretanto, os RVs evoluíram mecanismos de escape: suas proteases (2A e 3C) clivam adaptadores da via de sinalização (MAVS, TRIF), reduzindo a produção de IFN-ß. Essa deficiência relativa permite maior replicação viral e inflamação descontrolada, explicando por que a resposta ao IFN-ß é importante para controlar a infecção, mas frequentemente insuficiente nos quadros clínicos.
Na asma os mecanismos de respostas antivirais nas exacerbações são considerados deficientes e ainda não são inteiramente compreendidos. No entanto, está bem determinado que são potencialmente causadas por respostas Th2 exacerbadas (IL-4, IL-5 e IL-13) o que não ocorre em controles normais.62,67 Outro fator confirmado por metanálises mostra produção deficiente de IFN (IFN-ß e IFN-λ), tanto em adultos como em crianças com asma atópica. O ambiente de inflamação alérgica do paciente asmático, caracterizado por níveis elevados de IL-4 e IL-13, cria um cenário de maior vulnerabilidade a infecções virais. A supressão da resposta de interferon facilita a replicação do RV, o que explica a maior frequência e gravidade das exacerbações virais nestes pacientes.68,69
Segue-se uma resposta imune adaptativa pelas células Th1, caracterizada pela produção de IFN-g e IL-12, conduzindo a forte resposta antiviral, rápido clearance do vírus com mínima inflamação. No entanto, em pacientes com asma, tanto a resposta inata como a adaptativa podem estar comprometidas, determinando necrose epitelial e liberação de mediadores inflamatórios e do vírus propriamente dito. O aumento da carga viral e de mediadores inflamatórios liberados pela necrose das células do epitélio resultam em inflamação brônquica e consequente exacerbação da asma (Figura 5). O epitélio das vias aéreas (onde o RV se replica) tem respostas imunes inatas fracas. Isso causa um desequilíbrio para respostas Th2, com inflamação tipo 2 exagerada. Por isso, infecções por RV afetam mais o trato respiratório inferior de asmáticos do que de pessoas saudáveis.70
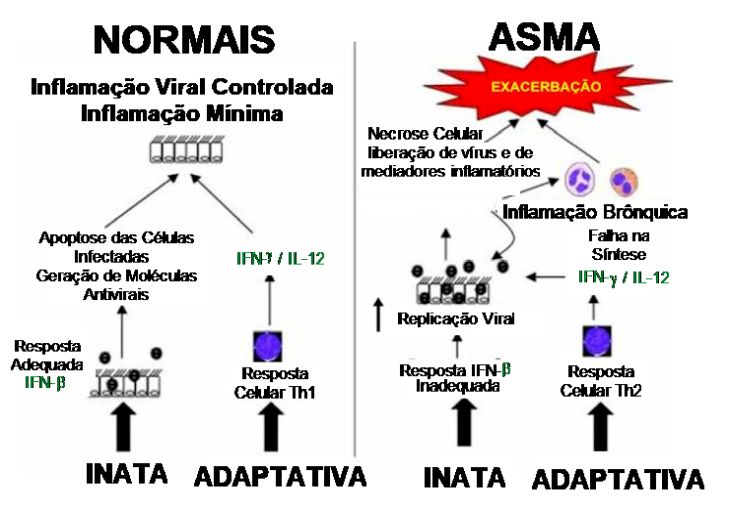
Figura 5 - Mecanismos da exacerbação da asma pela infecção viral. Retirado e adaptado de Mallia P. Johnston SL. Chest 2006; 130:1203-1210.
O IFN-β é a citocina responsável por iniciar a apoptose e o clearance viral pelas células epiteliais bem como por ativar vias protetoras antivirais. A produção do IFN-γ está também reduzida no epitélio de pacientes com asma e nos monócitos/macrófagos com infecção viral, sugerindo que uma deficiência genérica ocorra na produção de IFN, determinando maior suscetibilidade das vias aéreas às infecções virais do resfriado comum.
Publicação de Wark et al.,63 da Universidade de Southampton, introduziu uma nova explicação para a maior suscetibilidade dos pacientes com asma para a infecção pelo RV. Estudaram a replicação do RV-16 e como as células epiteliais respondem à infecção. A avaliação das respostas imunes inatas precoces, através de cultura, revelou profundo comprometimento na expressão do mRNA interferon-ß vírus-induzida. Os resultados evidenciaram que os pacientes com asma têm expressão RNA viral no sobrenadante 50 vezes maior quando comparado a voluntários sem asma, os quais estavam quase que completamente protegidos da infecção brônquica. Esta constatação é decorrente de uma falha na síntese de IFN-ß pelas células epiteliais brônquicas de asmáticos, > 2,5 vezes menos IFN-ß do que as de voluntários normais. Em células epiteliais infectadas de pacientes com asma, o IFN-ß exógeno induziu à apoptose, um dos mecanismos de defesa contra a infecção viral, e reduziu a replicação viral, demonstrando um link causal entre deficiência de IFN-ß, apoptose inadequada e aumento na replicação viral.
A grande maioria dos sorotipos do rinovírus humano (~160) utiliza a ICAM-1 como sítio de fixação nas células  susceptíveis, para iniciar a infecção. Uma minoria de sorotipos (1A, 1B, 2) se fixa sobre o receptor de lipoproteína de baixa densidade (LDL-R),
bem como CDHR3 para RV-C.71 A ICAM-1 é considerada na atualidade o maior receptor humano para o rinovírus (Figura 6). ICAM-1 é membro da superfamília das imunoglobulinas e é conhecida por desempenhar um importante papel nos processos de adesão celular na resposta imune. A upregulation da ICAM-1 tem sido detectada após infecção experimental com rinovírus. A ligação do vírus à ICAM-1 em diferentes tipos de células determina a ativação de vários tipos de citocinas e, além disso, a maior expressão da própria ICAM-1 em células adjacentes, o que favorece a adesão e disseminação do vírus. Com a maior expressão da ICAM-1, ocorre aumento da atração, migração e adesão de eosinófilos e neutrófilos, intensificando a inflamação com exacerbação da hiper-responsividade brônquica. Os níveis de exressão de ICAM-1 dos sorotipos RV-A e todos RV-B, podem ser regulados positivamente por estímulos ambientais, como poluentes atmosféricos e fumaça de cigarro.72 Essas condições podem aumentar a adesão, a entrada e replicação viral. susceptíveis, para iniciar a infecção. Uma minoria de sorotipos (1A, 1B, 2) se fixa sobre o receptor de lipoproteína de baixa densidade (LDL-R),
bem como CDHR3 para RV-C.71 A ICAM-1 é considerada na atualidade o maior receptor humano para o rinovírus (Figura 6). ICAM-1 é membro da superfamília das imunoglobulinas e é conhecida por desempenhar um importante papel nos processos de adesão celular na resposta imune. A upregulation da ICAM-1 tem sido detectada após infecção experimental com rinovírus. A ligação do vírus à ICAM-1 em diferentes tipos de células determina a ativação de vários tipos de citocinas e, além disso, a maior expressão da própria ICAM-1 em células adjacentes, o que favorece a adesão e disseminação do vírus. Com a maior expressão da ICAM-1, ocorre aumento da atração, migração e adesão de eosinófilos e neutrófilos, intensificando a inflamação com exacerbação da hiper-responsividade brônquica. Os níveis de exressão de ICAM-1 dos sorotipos RV-A e todos RV-B, podem ser regulados positivamente por estímulos ambientais, como poluentes atmosféricos e fumaça de cigarro.72 Essas condições podem aumentar a adesão, a entrada e replicação viral.
A necrose e anormalidades funcionais ciliares do epitélio proporcionam maior facilidade na penetração de alérgenos e/ou irritantes, determinando sensibilização alergênica, expondo também fibras nervosas. A exposição de fibras colinérgicas causa broncoconstrição resultante de uma disfunção dos receptores muscarínicos M273,74 e aumento da atividade eferente vagal e liberação de taquicininas (SP e NkA). Os neuropeptídios SP e NkA podem também contribuir para a obstrução brônquica através do aumento da síntese de leucotrienos, ativação de mediadores do mastócito e aumento da secreção de muco. A bradicinina, um polipeptídio com nove aminoácidos, é gerada a partir de precursores plasmáticos como parte do processo inflamatório, sendo encontrada nas secreções nasais de indivíduos infectados com o RV.
Alguns vírus podem determinar dano mediado via complemento (C). Componentes do complemento se ligam às células epiteliais tanto in vitro como in vivo, durante a infecção pelo VSR. C3a e C5a encontram-se aumentados em humanos voluntários infectados pelo vírus Influenza A.
Disfunção de receptores ß2-adrenérgicos pode também ocorrer por mediação viral75 Alterações do tono muscular podem estar relacionadas à perda de substâncias protetoras expressas ou produzidas pelo epitélio infectado, como o Fator Relaxante Derivado do Epitélio (EpDIF) e a Endopeptidase Neutra (NEP), uma enzima que degrada neuropeptídios.76
A infecção viral desencadeia resposta imune celular. Monócitos e macrófagos exibem uma intensa resposta antiviral, com produção de citocinas pró-inflamatórias (IL-1ß, IL-6, interferons e TNF-a) e eicosanoides.77,78 Os linfócitos T aumentam em número nas mucosas nasal e brônquica, coincidindo com linfopenia periférica, são ativados e produzem, em circunstâncias normais, o interferon-g que é a principal citocina secretada pelos linfócitos em resposta às infecções virais.79 Foi demonstrado que os linfócitos T CD8+em ambiente alérgico respondem à infecção viral produzindo IL-5 que por sua vez atrai os eosinófilos, ativando-os. Basófilos e mastócitos ativados produzem mediadores que incrementam também o recrutamento de eosinófilos nas vias aéreas, intensificando-se os efeitos de citotoxicidade e inflamação.
Na presença de infecção viral por RV, as células do epitélio nasal são as primeiras a serem afetadas. Secreção nasal de crianças com asma induzida por vírus evidenciou níveis aumentados de mieloperoxidase do neutrófilo, MBP, IL-8, RANTES e MIPa,80,.81 citocinas que são quimiotáxicas para linfócitos e eosinófilos. A replicação dos rinovírus em nível brônquico está relacionada a um aumento de citocinas que persiste por seis dias.
Vários estudos têm demonstrado aumento da hiper-responsividade brônquica (HRB) após infecções respiratórias. Infecções experimentais em humanos com RV aumentam a HRB a estímulos não específicos por mais de 4 semanas após a infecção em pacientes alérgicos.82 Mais recentemente, um estudo coorte em crianças com asma pós-viral foi prospectivamente acompanhado e a HRB periodicamente avaliada após o resfriado. A maioria das infecções documentadas era decorrente do RV (82%). A duração da HRB pós-viral é atualmente longa, ~ 7 semanas, quando subsequentes resfriados não ocorrem. Entretanto, quando de infecções recorrentes a HRB se prolonga por meses. Indivíduos atópicos habitualmente têm aumento no número de resfriados e, em consequência, prolongados períodos de maior HRB.83
O epitélio medeia processos inflamatórios complexos em resposta à exposição a alérgenos ou gatilhos não alérgicos, como os vírus, incluindo a liberação de um trio de citocinas epiteliais, conhecidas como "alarminas".84 As alarminas são moléculas liberadas rapidamente por células epiteliais ou imunes quando sofrem estresse, dano ou infecção. Em infecções virais elas têm papel decisivo como gatilhos iniciais da resposta imune inata. As alarminas IL-25, IL-33 e Linfopoetina Estromal Tímica (TSLP) estimulam respostas inflamatórias por meio de inúmeras vias downstream, incluindo endótipo tipo 2 (IL-4, IL-13 e IL-5) e outros, como vias conduzidas por Th1 ou Th17 (IL-17), resultando em vários desdobramentos fisiopatológicos que podem levar na asma a resposta imunológica amplificada e desregulada.84,85 Essa cascata inflamatória desencadeada pelas alarminas explica a exacerbação aguda dos sintomas durante infecção viral que ocorre na asma.
Beale et al.,86 em um modelo de infecção humana, demonstraram que o rinovírus era capaz de induzir a IL-25 no trato respiratório. Esta foi claramente menos potente em indivíduos saudáveis, enquanto que os níveis basais e de pico de infecção foram consideravelmente maiores na asma, demonstrando que as vias aéreas asmáticas possuíam propensão significativamente maior para produzir essa citocina. Os resultados da pesquisa suportam um mecanismo pelo qual o RV regula a expressão de IL-25 por células epiteliais de vias aéreas asmáticas suscetíveis. A IL-25 aumentada pode amplificar intensamente a cascata imune do tipo 2 envolvendo a ativação de células não T tipo 2 e células Th2 que expressam o receptor de IL-25.86
Jackson et al.87 em modelo experimental humano demonstraram que a infecção pelo RV leva à indução de IL-33 e citocinas tipo 2 na asma in vivo e que os níveis desses mediadores estão relacionados à gravidade das exacerbações da asma. Este foi o primeiro estudo experimental a mostrar que a infecção por RV do epitélio brônquico pode ativar diretamente células T humanas e células linfoides inatas (ILC2s) para produzir grandes quantidades de citocinas do tipo 2 (IL-5, IL-13), um processo completamente dependente de IL-33.87
Níveis elevados de TSLP em indivíduos asmáticos também se correlacionam positivamente com a obstrução das vias aéreas, uma característica que acaba levando à dificuldade respiratória.88-90 Em um estudo de células epiteliais nasais, a expressão de mRNA de TSLP estava significativamente aumentada pela estimulação do ligante TLR3, um receptor importante no reconhecimento de RNAs de fita dupla (dsRNAs) que ocorrem durante a replicação viral e que essa resposta era suprimida pelo tratamento com corticoides.91 Perez et al. identificaram que crianças de ≤ 3 anos com infecção por rinovírus apresentaram níveis médios nasal de TSLP ± SD mais altos em comparação com indivíduos da mesma idade sem nenhum vírus identificável (16,7 ± 1,2 pg·mL-1 versus 5,5 ± 0,9 pg·mL-1 – p<0,01).92
Tratamento
O tratamento da bronquiolite aguda pelo VSR em crianças varia conforme a gravidade: leve ⟹ (ambulatorial) moderada ⟹ (internação em enfermaria se houver risco) ⟹ grave (UTI).
Nenhuma medicação específica somente medidas de suporte, se necessário: oxigênio, hidratação e manejo de secreções. Broncodilatadores, corticoides e antibióticos não são recomendados de rotina. Antivirais (Ribavirina) somente em casos excepcionais.
Quanto à prevenção recomenda-se vacinação em gestantes entre 32–36 semanas com o objetivo de transferência de anticorpos transplacentários para proteger o lactente nos primeiros 6 meses de vida.93
Outra estratégia seria a imunização passiva somente em grupo de alto risco, mensal com Palivizumabe.94 Também se pode lançar mão do
Nirsevimabe, anticorpo monoclomal de dose única sazonal, que age bloqueando a replicação do vírus e prevenindo o agravamento da doença nos seis meses subsequentes ao seu uso. Já se encontra aprovado em vários países, inclusive no Brasil desde 2023 para todos os lactentes na temporada VSR95 e em todas as crianças abaixo de 24 meses de idade, vulneráveis à doença grave pelo vírus (prematuros com doença pulmonar crônica, cardiopatas congênitas, imunodeprimidos) com alta efetividade.
Duas vacinas de proteína F estabilizadas pré-fusão contra o VSR apresentaram resultados, mostrando alta eficácia na prevenção de doenças do trato respiratório inferior relacionadas ao VSR em adultos ≥60 anos.96,97 Apesar de as infecções por VSR em adultos muitas vezes se resolverem sem complicações, existe particular atenção para um segmento de alto risco em indivíduos idosos com várias condições crônicas, dentre elas a asma e a DPOC, quando a infecção pelo VSR pode desencadear exacerbações graves, resultando em insuficiência respiratória, hospitalização prolongada e até mesmo morte.98 O VSR foi responsável por 11,4% das hospitalizações dos pacientes com DPOC e por 7,2% das hospitalizações de pacientes com asma, destacando-se como importante agente etiológico na descompesação dessas doenças.99
O vírus da influenza é passível de tratamento preventivo, através de vacina que é considerada eficaz. A eficácia da vacina varia a cada ano e não se aplica a cepas panendêmicas. Em adultos jovens saudáveis, a eficácia da vacina influenza é de cerca de 70 a 90% e em idosos alcança 60%.46 A vacina contra a gripe deve ser administrada antes que o vírus comece a circular na comunidade, pois leva cerca de duas semanas após a vacinação, para que os anticorpos se desenvolvam no corpo e forneçam proteção contra a gripe.
Para o tratamento da gripe influenza em pacientes com asma existem dois medicamentos que reduzem sintomas, mas o efeito em prevenir ou reduzir exacerbações de asma é limitado para ambos: o Oseltamivir e o Baloxavir. O estudo de Ison et al.100 em uma população de risco que incluiu asmáticos, utilizando o Baloxavir, que se usa em dose única, apresentou eficácia superior ao placebo e semelhante ao Oseltamivir na melhora dos sintomas em população de alto risco. Propiciou redução do tempo de eliminação viral. O estudo incluiu subtipo de pacientes com asma, mas não foi desenhado com poder estatístico exclusivo para desfechos de exacerbação asmática.100 Por outro lado, o oseltamivir em crianças asmáticas melhora a função pulmonar e reduz a frequência de exacerbações,101 e quando indicado precocemente reduz complicações respiratórias.102
Atualmente não há tratamento eficaz que proteja contra a infecção do RV e limite a infecção e progressão da doença. Várias substâncias com ação antiviral têm sido avaliadas contra o RV, com resultados pouco animadores. Dentre elas, o ácido ascórbico, o zinco, fragmentos do receptor VLDL, derivados solúveis da ICAM. Entre as primeiras estratégias antivirais, os medicamentos de ligação ao capsídeo direcionados ao vírus (Pleconaril, Pirodavir, Vapendavir) mostraram eficácia, impedindo a entrada do vírus,103,104 entretanto, desenvolveram resistência rapidamente aos medicamentos.105-107
A busca por uma vacina contra o RV permanece sem sucesso após décadas de estudo, principalmente devido à sua grande diversidade antigênica e à incapacidade de induzir resposta imune duradoura e de proteção cruzada. No entanto, o cenário começa a mudar com avanços recentes. A identificação de epítopos neutralizantes conservados, o desenvolvimento de vacinas candidatas polivalentes e específicas (como para o RV-C), e os novos conhecimentos sobre a estrutura viral e sua interação com receptores estão reacendendo o otimismo e revitalizando os esforços de desenvolvimento.71,108,109
Os IFNs são outra classe de moléculas imunoestimulantes inatas antivirais que foram largamente estudados. IFN-β nebulizado inalado foi testado em uma coorte de asmáticos para determinar se pode prevenir exacerbações virais. Apesar de não atingir seu desfecho primário, esta pesquisa mostrou o potencial de modular as respostas imunes inatas em populações susceptíveis.110,111 Pode ser mais eficaz se administrado nos primeiros sinais de infecção (profilaxia pós-exposição ou tratamento muito precoce), e não após a exacerbação já estar estabelecida.
Pode beneficiar apenas um subtipo específico de pacientes asmáticos (p. ex. aqueles com deficiência mais pronunciada de interferon, com certos fenótipos ou gravidade).
Não há, atualmente, nenhuma indicação aprovada para o uso rotineiro do IFN-β no tratamento ou profilaxia de infecções por rinovírus em pacientes asmáticos.
Referências
1.Rabinowitz HK. Upper respiratory tract infections. Prim Care 1990; 17:793-809.
2.Johnston SL. – Viral infections in children wiTh existing asThma. In: From Genetics to Quality of Life. Seattle. Hogrefe & Huber Publishers, 1996.
3.Podosin RL, Felton WL. The clinical picture of a Far-East influenza occurring at The 4Th National Boy Scout Jamboree. N.Engl J Med 1958; 238:778-82.
4.Rebhan AW. An outbreak of Asian influenza in a girls'camp. Can Med Assoc J 1997; 77:797-9.
5.Noah TL, Henderson FW, Wortman IA, Devlin RB, Handy J, Koren HS, Becker S.
Nasal cytokine production in viral upper respiratory infection of childhood. J Infect Dis 1995; 171:584-92.
6.Turner RB. The common cold. Pediatr Ann 1998; 27:790-5.
7.Adams PF, Hendershot GE, Marano MA; Centers for Disease Control and Prevention/National Center for HealTh Statistics. Current estimates from The National HealTh Interview Survey, 1996. Vital HealTh Stat 10. 1999: 1-203.
8.Fendrick AM, Monto AS, Nightengale B, Sarnes M. The economic burden of non-influenza-related viral respiratory tract infection in The United States. Arch Intern Med 2003; 163:487-94.
9.Openshaw PJ, Lemanske, RF. Respiratory viruses and asThma:can The effects be prevented? Eur Respir J 1998; 12:suppl.27:35s-39s.
10.Bardin PG, Fraenkel D, Sanderson G, Dorward M, Johnston S, Holgate S.
Increased sensitivity to The consequences of rhinoviral infection in atopic subjects. Chest 1995; 107(Suppl.3):157S.
11.Micillo E, Marcatili P, Palmieri S, Mazzarella G. Viruses and asThmatic syndromes. Monaldi Arch Chest Dis 1998; 53:88-91.
12.Johnston SL. Natural and experimental infection of lower respiratory tract. Am J Respir Crit Care Med 1995; 152:S46-52.
13.Pattemore PK, Johnston SL, Bardin PG. Viruses as precipitants of asThma symptoms. I. Epidemiology. Clin Exp Allergy 1992; 22:325-36.
14.Horn MEC. Brain EA, Gregg I, Inglis JM, Yealland SJ, Taylor P. Respiratory viral infection and wheezy bronchitis in childhood. Thorax 1979; 34:23-8.
15.Hudgel DW, Langston LJ, Selner JC, McIntosh K. Viral and bacterial infections in adults wiTh chronic asThma. Am Rev Respir Dis 1979; 120:393-7.
16.Jennings LC, Barns G, Dawson KP. The association of viruses wiTh acute asThma. NZ Med J 1987; 100:488-90.
17.Cheung D, Dick EC, Timmers MC, de Klerk EPA, Spaan WJM, Sterk PJ. Rhinovirus inhalation causes long-lasting excessive airway narrowing in response to meThacholine in asThmatic subjects in vivo. Am J Respir Crit Care Med 1995; 152:1490-6.
18.Lemanske RF, Dick EC, Swenson CA, Vrtis RF, Busse WW. Rhinovirus upper respiratory infection increases airway hyperreactivity and late asThmatic reactions. J Clin Invest 1989; 83:1-10.
19.Arruda E, Pitkäranta A, Witek TJ Jr, Doyle CA, Hayden FG. Frequency and natural history of rhinovirus infections in adults during autumn. J Clin Microbiol 1997; 35:2864-68.
20.Nicholson KG, Kent J, Hammersley V, Cancio E.
Risk factors for lower respiratory complications of rhinovirus infections for elderly people living in The community: prospective cohort study. BMJ 1996; 313:1119-23.
21.Johnston SL, Pattemore PK, Sanderson G, SmiTh S, Lampe F, Josephs L, Sympington P, O'Toole S, Myint SH, Tyrrell DA, Holgate ST. Community study of role of viral infections in exacerbations of asThma in 9-11 year old children. Br Med J 1995; 310:1225-9.
22.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asThma in adults. BMJ 1993; 307:982-6.
23.Wright AL, Holberg CJ, Martinez FD, Morgan WJ, Taussig LM. Breast feeding and lower respiratory tract illness in The first year of life. Br Med J 1989; 299:946-9.
24.EfstaThiou C, Abidi SH, Harker J, Stevenson NJ. Revisiting respiratory syncytial virus's interaction wiTh host immunity, towards novel Therapeutics. Cell Mol Life Sci 2020; 77:5045-58.
25.Dalziel SR, Haskell L, O'Brien S, Borland ML, Plint AC, Babl FE, Oakley E. Bronchiolitis. Lancet 2022; 400(10349):392-406. doi: 10.1016/S0140-6736(22)01016-9.
26.Fujiogi M, Goto T, Yasunaga H, Fujishiro J, Mansbach JM, Camargo CA Jr, Hasegawa K. Trends in Bronchiolitis Hospitalizations in The United States: 2000-2016. Pediatrics 2019; 144:e20192614. doi: 10.1542/peds.2019-2614.
27.Hasegawa K, Dumas O, Hartert TV, Camargo CA Jr. Advancing our understanding of infant bronchiolitis Through phenotyping and endotyping: clinical and molecular approaches. Expert Rev Respir Med 2016; 10:891-9.
28.Régnier SA, Huels J. Association between respiratory syncytial vírus hospitalizations in infants and respiratory sequelae: systematic review and meta-analysis. Pediatr Infect Dis J 2013; 32:820-6.
29.Liu L, Pan Y, Zhu Y, Song Y, Su X, Yang L, Li M.
Association between rhinovirus wheezing Illnes and The development of childhood asThma: a meta-analysis.
BMJ Open. 2017 Apr 3;7(4):e013034. doi: 10.1136/bmjopen-2016-013034.
30.Fujiogi M, Dumas O, Hasegawa K, Jartti T, Camargo CA. Identifying and predicting severe bronchiolitis profiles at high risk for developing asThma: Analysis of Three prospective cohorts. eClinicalMedicine 2022;43: 101257 doi:https://doi.org/10.1016/j.eclinm.2021.101257.
31.Korppi M, Törmänen S. Toll-like receptor 1 and 10 variations increase asThma risk and review highlights furTher research directions. Acta Paediatr 2019; 108:1406-10.
32.Duff AL, Pomeranz ES, Gelber LE, Price GW, Farris H, Hayden FG, Platts-Mills TAE, Heymann RW. Risk factors in acute wheezing in infants and children: viruses, passive smoke, and IgE antibodies to inhalant allergens. Peds 1993; 92:535-40.
33.Rotbart HA, Hayden FG. Picornavirus infections: a primer for practitioner. Arch Fam Med 2000; 9:913. 34. Temte JL. A family physician's perspective on picornavirus infections in primary care. Arch Fam Med 2000; 9:921-2.
34.Semler BL, Ertel KJ. Picornaviruses: Molecular Biology. In: Brian W.J. Mahy & Marc H.V. van Regenmortel. Encyclopedia of Virology. Academic Press; 2008:129-140.
35.Palmenberg AC. – Sequence alignments of picornaviral capsid proteins. In: Semler BL, Ehrenfeld E. Molecular Aspects of Picornavirus Infection and Detection. Washington. DC: American Society of Microbiology; 1989:211-241.
36.The Pirbright Institute, UK. Disponível em: https://www.picornaviridae.com/ Acesso em 20 de julho de 2022.
37.Picornavirus Home. Picornaviridae Study Group. Disponível em: https://www.picornastudygroup.com/ Acesso em 20 de julho de 2022.
38.Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, Martin AC, Khoo SK, Cox DW, Geelhoed GC, McMinn PC, Goldblatt J, Gern JE, Le Souëf PN. Association between human rhinovirus C and severity of acute asThma in children. Eur Respir J 2011; 37:1037-42.
39.Price AS, Kennedy JL. T-helper 2 mechanisms involved in human rhinovirus infections and asThma. Ann Allergy AsThma Immunol 2022; 129:681-691.
40.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, Holgate ST, Davies DE. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med 2005; 201:937-47.
41.Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, Kebadze T, Mallia P, Stanciu LA, Parker HL, Slater L, Lewis-Antes A, Kon OM, Holgate ST, Davies DE, Kotenko SV, Papi A, Johnston SL. Role of deficient type III interferon-lambda production in asThma exacerbations. Nat Med 2006; 12:10231026.
42.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, Contoli M, Sanderson G, Kon OM, Papi A, Jeffery PK, Stanciu LA, Johnston SL. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci U S A 2008; 105:13562-7.
43.Sykes A, Edwards MR, Macintyre J, del Rosario A, Bakhsoliani E, Trujillo-Torralbo MB, Kon OM, Mallia P, McHale M, Johnston SL. Rhinovirus 16-induced IFN-a and IFN-ß are deficient in bronchoalveolar lavage cells in asthmatic patients. J Allergy Clin Immunol 2012; 129:1506-1514.e6.
44.Baraldo S, Contoli M, Bazzan E, Turato G, Padovani A, Marku B, Calabrese F, Caramori G, Ballarin A, Snijders D, Barbato A, Saetta M, Papi A. Deficient antiviral immune responses in childhood: distinct roles of atopy and asThma. J Allergy Clin Immunol 2012; 130:1307-14.
45.Dowdle WR, Noble GR, Kendal AP. – Orthomyxovirus-influenza: Comparative diagnosis unifiying concept. In: Krustak C. Comparative Diagnosis of Viral Diseases. New York: Academic Press: 1977:447.
46.Guia de Imunizações SBIm/SBPT Pneumologia 2019/2019 Disponível em: https://www.sbpt.org.br
47.Williams JV, Crowe JE Jr, Enriquez R, et al. Human metapneumovirus infection plays an etiologic role in acute asThma exacerbations requiring hospitalization in adults. J Infect Dis 2005; 192:1149-53.
48.Xepapadaki P, Psarras S, Bossios A. Human metapneumovirus as a causative agent of acute bronchiolitis in infants. J Clin Virol 2004; 30:267-70.
49.Pifferi M, Maggi F, Andreoli E, at al. Associations between nasal torquetenovirus load and spirometric indices in children wiTh asThma. J Infect Dis 2005; 192:1141-8.
50.Proud D, Gwaltney JM Jr, Hendley JO, Dinarello CA, Gillis S, Schleimer RP. Increased levels of interleukin-1 are detected in nasal secretions of volunteers during experimental rhinovirus colds. J Infect Dis 1994; 169:1007-13.
51.Zhu Z, Tang W, Gwaltney JM Jr, Wu Y, Elias JA. Rhinovirus stimulation of interleukin-8 in vivo and in vitro:role of NF-kB. Am J Physiol 1997; 273:L814-24.
52.Zhu Z, Tang W, Ray A, Wu Y, Einarsson O, Landry ML, Gwaltney JM Jr, Elias JA. Rhinovirus stimulation of interleukin-6 in vivo and in vitro. Evidence for nuclear factor kB-dependent transcriptional activation. J Clin Invest 1996; 97:421-30.
53.Rot A, Krieger M, Brunner T, Bischoff SC, Schall TJ, Dahinden CA. RANTES and macrophage inflammatory protein 1a induce The migration and activation of normal human eosinophil granulocytes. J Exp Med 1992; 176:1489-95.
54.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of The memory phenotype by cytokine RANTES. Nature 1990; 347:669-71.
55.Garcia-Zepeda EA, RoThenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nature Med 1996; 2:449-56.
56.PonaTh PD, Qin S, Ringler DJ, Clark-Lewis I, Wang J, Kassam N, SmiTh H, Shi X, Gonzalo JA, Newman W, Guiterrez-Ramos JC, Mackay CR. Cloning of human eosinophil chemoattractant, eotaxin. J Clin Ivest 1996; 97:604-12.
57.Donninger H, Glashoff R, Haitchi HM, Syce JA, Ghildyal R, van Rensburg E, Bardin PG.
Rhinovirus induction of The CXC chemokine epiThelial-neutrophil activating peptide-78 in bronchial epiThelium. J Infect Dis 2003; 187:1809-17.
58.Vignola AM, Campbell AM, Chanez P, Bousquet J, Paul-Lacoste P, Michel FB, Godard P. HLA-DR and ICAM-1 expression on bronchial epiThelial cells in asThma and chronic bronchitis. Am Rev Respir Dis 1993; 148:689-94.
59.Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a human respiratory epiThelial cell line wiTh rhinovirus. J Clin Invest 1995; 96:549-57.
60.Psarras S. Rhinovirus infection stimulates production of fibroblast growTh factor-2 by airway epiThelial and stromal cells. Allergy 2002; 57:suppl. 73.
61.McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol 2015; 15:87-103.
62.Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, Jerico Del-Rosario, Telcian AG, Nikonova A, Zhu J, Aniscenko J, Gogsadze L, Bakhsoliani E, Traub S, Dhariwal J, Porter J, Hunt D, Hunt T, Hunt T, Stanciu LA, Khaitov M, Bartlett NW, Edwards MR, Kon OM, Mallia P, Papadopoulos NG, Akdis CA, Westwick J, Edwards MJ, Cousins DJ, Walton RP, Johnston SL. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med 2014; 190:1373-82.
63.Wark PAB, Johnston SL, Bucchieri F, Powell R., et.al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med 2005; 201:937947.
64.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 2014; 32:513-45.
65.Aaronson, D. S., & Horvath, C. M. (2002). A roadmap of the JAK - STAT signaling pathway. Science STKE, 2002 (117), re6.
66.Yang Z, Mitländer H, Vuorinen T, Finotto S. Mechanism of Rhinovirus Immunity and Asthma. Front Immunol 2021; 12:731846. doi: 10.3389/fimmu.2021.731846. Erratum in: Front Immunol 2021; 12:800020.
67.Farne H, Glanville N, Johnson N, Kebadze T, Aniscenko J, Regis E, Zhu J, Trujillo-Torralbo MB, Kon OM, Mallia P, Prevost AT, Edwards MR, Johnston SL, Singanayagam A, Jackson DJ. Effect of CRTH2 antagonism on the response to experimental rhinovirus infection in asthma: a pilot randomised controlled trial. Thorax 2022t; 77:950-959.
68.Contoli M, Ito K, Padovani A, Poletti D, Marku B, Edwards MR, Stanciu LA, Gnesini G, Pastore A, Spanevello A, Morelli P, Johnston SL, Caramori G, Papi A. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy 2015; 70:910-20.
69.Gielen V, Sykes A, Zhu J, Chan B, Macintyre J, Regamey N, Kieninger E, Gupta A, Shoemark A, Bossley C, Davies J, Saglani S, Walker P, Nicholson SE, Dalpke AH, Kon OM, Bush A, Johnston SL, Edwards MR. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J Allergy Clin Immunol 2015l; 136:177-188.e11.
70.
Mallia P, Johnston SL. How viral infections cause exacerbation of airway diseases. Chest 2006; 130:1203-10.
71.Lee E, Gern JE. Clinical Significance of Rhinoviruses and Progress Toward Vaccination. Allergy Asthma Immunol Res 2025l; 17:414-432.
72.Shukla SD, Mahmood MQ, Weston S, Latham R, Muller HK, Sohal SS, Walters EH. The main rhinovirus respiratory tract adhesion site (ICAM-1) is upregulated in smokers and patients with chronic airflow limitation (CAL). Respir Res 2017; 18:6.
73.Fryer A, Yarkony K, Jacoby D. The effect of leukocyte depletion on pulmonary M2 muscarinic receptor function in parainfluenza virus infected guinea pigs. Br J Pharmacol 1994; 112:588-94.
74.Fryer A, Jacoby D. Parainfluenza virus infection damages inhibitory M2 muscarinic receptors on pulmonary parasympaThetic nerves in The guinea-pig. Br J Pharmacol 1991; 102:267-71.
75.Buckner CK, Clayton DE, Ain-Shoka AA, Busse WW, Dick EC, Shult P. Parainfluenza 3 infection blocks The ability of a beta-adrenergic receptor agonist to inhibit antigen-induced contraction of guinea pig isolated-induced airway smooTh muscle. J Clin Invest 1981; 67:376-84.
76.Nadel JA, Borson DB. Modulation of neurogenic inflammation by neutral endopeptidase. Am Rev Respir Dis 1991; 143:S33-6.
77.Hegele RG, Hayashi S, Hogg JC, Pare PD. Mechanisms of airway narrowing and hyperresponsiveness in viral respiratory tract infections. Am J Respir Crit Care Med 1995; 151:1659-64.
78.Peschke T, Bender A, Naim M, Gemsa D. Role of macrophage cytokines in influenza A virus infections. Immunobiology 1993; 189:340-55.
79.Noah T, Henderson F, Wortman I et al. Nasal cytokine production in viral upper respiratory infection of childhood. J Infect Dis 1995; 171:584-92.
80.Seminario M-C, Squillace D, Bardin PG, Fraenkel DJ, Gleich GJ, Johnston SL. Increased levels of eosinophil major basic protein in nasal secretions in rhinovirus infection. J Allergy Clin Immunol 1995; 95:259.
[ Google Scholar ]
81.Teran LM, Johnston SL, Shute JK, Church MK, Holgate ST. Increased levels of interleukin-8 in The nasal aspirates of children wiTh virus-associated asThma. J Allergy Clin Immunol 1994; 93:272.
82.Gern JE, Calhoun W, Swenson C, Shen G, Busse WW. Rhinovirus infection preferentially increases lower airway responsiveness in allergic subjects. Am J Respir Crit Care Med 1997; 155:1872-76.
83.Xepapadaki P, Papadopoulos NG, Bossios A, Manoussakis E, Manousakas T, Saxoni-Papageorgiou P. Duration of postviral airway hyperresponsiveness in children wiTh asThma: effect os atopy. J Allergy Clin Immunol 2005; 116:299-304.
84.Mitchell PD, O'Byrne PM. EpiThelial-derived cytokines in asThma. Chest 2017;151:13381344.
85.Porsbjerg CM, Sverrild A, Lloyd CM, et al. Anti-alarmins in asThma: targeting The airway epiThelium wiTh next-generation biologics. Eur Respir J 2020; 56:2000260 [https://doi.org/10.1183/ 13993003.00260-2020].
86.Beale J, Jayaraman A, Jackson DJ, Macintyre JDR, Edwards MR, Walton RP, Zhu J, Man Ching Y, Shamji B, Edwards M, Westwick J, Cousins DJ, Yi Hwang Y, McKenzie A, Johnston SL, Bartlett NW. Rhinovirus-induced IL-25 in asThma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med 2014; 6:256ra134.
87.Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, Jerico Del-Rosario, Telcian AG, Nikonova A, Zhu J, Aniscenko J, Gogsadze L, Bakhsoliani E, Traub S, Dhariwal J, Porter J, Hunt D, Hunt T, Hunt T, Stanciu LA, Khaitov M, Bartlett NW, Edwards MR, Kon OM, Mallia P, Papadopoulos NG, Akdis CA, Westwick J, Edwards MJ, Cousins DJ, Walton RP, Johnston SL. IL-33-dependent type 2 inflammation during rhinovirus-induced asThma exacerbations in vivo. Am J Respir Crit Care Med 2014; 190:1373-82.
88.Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee Th, Corrigan C.
Thymic stromal lymphopoietin expression is increased in asThmatic airways and correlates wiTh expression of Th2-attracting chemokines and disease severity. J Immunol 2005; 174:8183-90.
89.Shikotra A, Choy DF, Ohri CM, Doran E, Butler C, Hargadon B, Shelley M, Abbas AR, Austin CD, Jackman J, Wu LC, Heaney LG, Arron JR, Bradding P.
Increased expression of immunoreactive Thymic stromal lymphopoietin in patients wiTh severe asThma. J Allergy Clin Immunol 2012; 129:104-11.e1-9.
90.Xu G, Zhang L, Wang DY, Xu R, Liu Z, Han DM, Wang XD, Zuo KJ, Li HB.
Opposing roles of IL-17A and IL-25 in The regulation of TSLP production in human nasal epiThelial cells. Allergy 2010; 65:581-9.
91.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of Thymic stromal lymphopoietin in human airway epiThelial cells. J Immunol 2007; 179:1080-7.
92.Perez GF, Pancham K, Huseni S, Preciado D, Freishtat RJ, Colberg-Poley AM, Hoffman EP, Rose MC, Nino G. Rhinovirus infection in young children is associated wiTh elevated airway TSLP levels. Eur Respir J 2014; 44:1075-8.
93.Patel D, Chawla J, Blavo C. Use of the Abrysvo Vaccine in Pregnancy to Prevent Respiratory Syncytial Virus in Infants: A Review. Cureus 2024; 16:e68349.
94.Yun KW. Recent Advances in the Prevention of RSV in Neonates and Young Infants. Pediatr Infect Vaccine 2023;30:111. 11.
95.Saúde incorpora vacina para proteger gestantes e bebês dos vírus sincicial respiratório. Ministério da Saúde, 2025. Disponível em: https://www.gov.br/saude/pt-br/assuntos/noticias/2025/fevereiro/saude-incorpora-vacina-para-proteger-gestantes-e-bebes-do-virus-sincicial-respiratorio.
96.Walsh EE, Pérez Marc G, Zareba AM, Falsey AR, Jiang Q, Patton M, Polack FP, Llapur C, Doreski PA, Ilangovan K, Rämet M, Fukushima Y, Hussen N, Bont LJ, Cardona J, DeHaan E, Castillo Villa G, Ingilizova M, Eiras D, Mikati T, Shah RN, Schneider K, Cooper D, Koury K, Lino MM, Anderson AS, Jansen KU, Swanson KA, Gurtman A, Gruber WC, Schmoele-Thoma B; RENOIR Clinical Trial Group. Efficacy and Safety of a Bivalent RSV Prefusion F Vaccine in Older Adults. N Engl J Med 2023; 388:1465-1477.
97.Papi A, Ison MG, Langley JM, Lee DG, Leroux-Roels I, Martinon-Torres F, Schwarz TF, van Zyl-Smit RN, Campora L, Dezutter N, de Schrevel N, Fissette L, David MP, Van der Wielen M, Kostanyan L, Hulstrøm V; AReSVi-006 Study Group. Respiratory Syncytial Virus Prefusion F Protein Vaccine in Older Adults. N Engl J Med 2023; 388:595-608.
98.Lee N, Lui GC, Wong KT, Li TC, Tse EC, Chan JY, Yu J, Wong SS, Choi KW, Wong RY, Ngai KL, Hui DS, Chan PK. High morbidity and mortality in adults hospitalized for respiratory syncytial virus infections. Clin Infect Dis 2013; 57:1069-77.
99.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 2005;352:1749-59.
100.Ison MG, Portsmouth S, Yoshida Y, Shishido T, Mitchener M, Tsuchiya K, Uehara T, Hayden FG. Early treatment with baloxavir marboxil in high-risk adolescent and adult outpatients with uncomplicated influenza (CAPSTONE-2): a randomised, placebo-controlled, phase 3 trial. Lancet Infect Dis 2020; 20:1204-1214.
101.Johnston SL, Ferrero F, Garcia ML, Dutkowski R. Oral oseltamivir improves pulmonary function and reduces exacerbation frequency for influenza-infected children with asthma. Pediatr Infect Dis J 2005; 24:225-32.
102.Uyeki TM. Oseltamivir Treatment of Influenza in Children. Clin Infect Dis 2018; 66:1501-1503.
103.Hayden FG, Herrington DT, Coats TL, Kim K, Cooper EC, Villano SA, Liu S, Hudson S, Pevear DC, Collett M, McKinlay M; Pleconaril Respiratory Infection Study Group. Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: results of 2 double-blind, randomized, placebo-controlled trials. Clin Infect Dis 2003; 36:1523-32.
104.Hayden FG, Andries K, Janssen PA. Safety and efficacy of intranasal pirodavir (R77975) in experimental rhinovirus infection. Antimicrob Agents ChemoTher 1992; 36:727-32.
105.Senior K. FDA panel rejects common cold treatment. Lancet Infect Dis 2002;2:264. doi: 10.1016/s1473-3099(02)00277-3.
106.Lanko K, Sun L, Froeyen M, Leyssen P, Delang L, Mirabelli C, Neyts J. Comparative analysis of The molecular mechanism of resistance to vapendavir across a panel of picornavirus species. Antiviral Res 2021; 195:105177. doi: 10.1016/j.antiviral.2021.105177.
107.Pevear DC, Hayden FG, Demenczuk TM, Barone LR, McKinlay MA, Collett MS. Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses. Antimicrob Agents ChemoTher 2005; 49:4492-9.
108.Pyle CJ , Patel ND , Edwards MR , et al. A Rhinovirus VP0 vaccine induces cross-reactive type 1 immunity and promotes lymphocyte activation and maturation (abstract). Am J Respir Crit Care Med 2024; 209:A6845.
109.Pyle CJ, Patel ND, Edwards MR, Shaw S, Johnston SL. Pre-clinical development of a novel cross-protective rhinovirus vaccin. Eur Respir J 2024; 64:Suppl.68,OA5461.
110.Djukanovic R, Harrison T, Johnston SL, Gabbay F, Wark P, Thomson NC, Niven R, Singh D, Reddel HK, Davies DE, Marsden R, Boxall C, Dudley S, Plagnol V, Holgate ST, Monk P; INTERCIA Study Group. The effect of inhaled IFN-ß on worsening of asThma symptoms caused by viral infections. A randomized trial. Am J Respir Crit Care Med 2014; 190:145-54.
111.Esneau C, Duff AC, Bartlett NW. Understanding Rhinovirus Circulation and Impact on Illness. Viruses 2022; 14:141. doi: 10.3390/v14010141.
|
|
Home
 Design by Walter Serralheiro Design by Walter Serralheiro
|
|
|