Os receptores esteroides, incluindo o GR humano, apresentam uma estrutura modular composta por cinco a seis regiões. A Figura 3 apresenta a representação linear do receptor GR humano com 777 aminoácidos (300 kDa) mostrando os domínios principais. Estão divididos em domínios funcionais assim denominados: domínio aminoterminal (DTN) – domínio de  transativação; domínio de ligação ao DNA (DBD) – DNA binding domain, região de dimerização do receptor; AF-1 e AF-2 – os dois domínios de ativação e região CT (carboxi-terminal) com domínio de ligação ao ligante (LBD – ligant binding domain) – regiões de localização nuclear.18
transativação; domínio de ligação ao DNA (DBD) – DNA binding domain, região de dimerização do receptor; AF-1 e AF-2 – os dois domínios de ativação e região CT (carboxi-terminal) com domínio de ligação ao ligante (LBD – ligant binding domain) – regiões de localização nuclear.18

Figura 3 — Representação da estrutura de gene do receptor glicocorticoide.
A representação linear do GR é constituída por 777 aminoácidos. O domínio aminoterminal (DTN) é codificado pelo éxon 2, composto por aproximadamente 439 aminoácidos e é o principal responsável pela maior parte da atividade transcricional independente do ligante, denominada função de ativação 1 (AF-1). É o sítio imunogenético do receptor e possui importante especificidade junto ao promotor. Este domínio contém a função de ativação (AF-1) transcricional constitutiva independente do ligante, que é necessária para a ativação transcricional máxima do GR.18-20 AF-1 é rico em aminoácidos ácidos e é importante para iniciação da transcrição, ou seja, interação de GR com correguladores, moduladores da cromatina e o mecanismo de transcrição basal.18,21,22
A região AF-1 contém 200 aminoácidos, com função de ativação transcricional hormônio independente.23 A atividade transcricional do AF-1 parece residir na região central de 41 aminoácidos carregados negativamente por aminoácidos ácidos.24 Tem-se demonstrado que esta região interage com fatores gerais de transcrição e com um número de proteínas coativadoras, apresentando, ainda, importante participação na repressão transcricional pelo GR.
Os próximos 65 aminoácidos na região central do GR compõem o DBD, codificado pelos éxons 3 e 4. Consiste em dois dedos de zinco altamente preservados.
Uma característica comum entre os dedos de zinco é a capacidade de reconhecer moléculas de DNA ou RNA de diferentes comprimentos, razão pela qual elas foram inicialmente consideradas apenas como fatores transcricionais. Esses aminoácidos são coordenados em tetraedro a um íon de zinco, mantido por quatro resíduos cisteínas. Cada unidade repetida com 30 aminoácidos é dobrada em um domínio alongado ligado ao zinco com a forma de um dedo, daí o nome dado de Zinc finger. O domínio de ligação ao DNA (DBD) é importante pela especificidade de ligação ao DNA, para a homodimerização do GR e interação com outros fatores de transcrição. O domínio DBD ocupa a região central do receptor e
contém aminoácidos responsáveis pela ligação de GR a GREs ( Elemento Responsivo aos Glicocorticoides). (Figura 4)
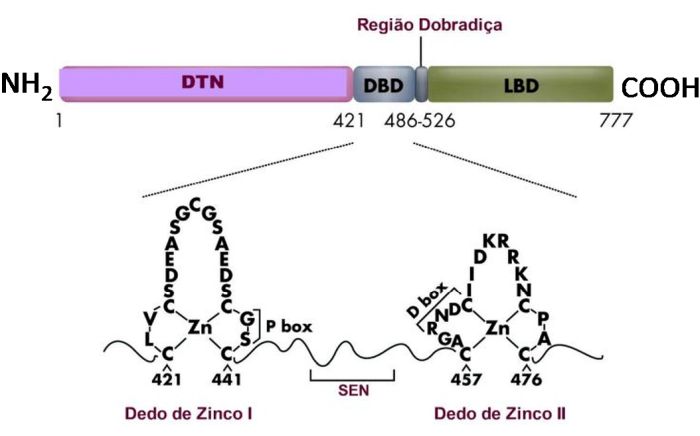
Figura 4 — Estrutura GR humano. O GR é composto por uma região DTN, uma DBD composta por dois dedos de zinco, uma região de dobradiça e uma região LBD C-terminal. A variação na função do GR pode ser correlacionada com esses domínios. P, proximal; D, distal. O DBD também contém a sequência de sinal de exportação nuclear (SEN) que o direciona para exportação do núcleo da célula para o citoplasma através do complexo de poros nucleares. Figura retirada e modificada de Vandevyver et al.18
Cada dedo de zinco é formado por cisteínas coordenadas a um íon de zinco (Zn2+) , formando uma estrutura que permite a interação com sequências específicas de DNA.
Os dois dedos de zinco são separados por uma sequência de 15–17 aminoácidos. Três aminoácidos da base do primeiro dedo representam a região chamada P-box
(ou "caixa de reconhecimento"), que reconhece e se liga às sequências específicas do DNA, conhecidas como elementos responsivos a glicocorticoides (GRE - Glucocorticoid Response Elements).
Esse dedo se liga ao sulco maior do DNA, garantindo o reconhecimento específico da sequência. Ao passo que uma sequência de cinco aminoácidos localizada entre a primeira e segunda cisteína na base do segundo dedo,
contém a D-box, envolvida na dimerização do receptor sobre o DNA, permitindo que dois receptores de glicocorticoide formem um dímero para ativar ou reprimir a transcrição gênica.15-27 Esse dedo contribui para a estabilidade da interação com o DNA.
A presença desses dois dedos de zinco no domínio DBD permite que o GR atue como um regulador da expressão gênica, modulando a transcrição de genes-alvo em resposta aos glicocorticoides.
Éxons 5-9 codificam a região da dobradiça, que separa o DBD do domínio de ligação do ligante (LBD). Abrange aminoácidos 486-526, é uma estrutura de ligação flexível que está implicada em permitir a ligação adequada do DNA, dimerização e translocação nuclear do receptor e
desempenha um papel crítico na ativação induzida pelo ligante do GRα.28 O LBD C-terminal contém o sítio de ligação do ligante e um segundo domínio de transativação (AF-2) regulado pela ligação hormonal.29
O domínio de transativação AF-2 é importante para a interação com cochaperonas, correguladores (chaperona – acompanhante, em inglês) e outros fatores de transcrição.30 O LBD também engloba uma interface de dímero que é crítica para a função GR e a ligação da proteína de choque térmico HSPs90.31 O DBD e o LBD contêm sinais de localização nuclear, que são importantes para a translocação nuclear GR. O DBD também contém a sequência de sinal de exportação nuclear (SEN) que o direciona para exportação do núcleo da célula para o citoplasma através do complexo de poros nucleares.32
➢ As cochaperonas são proteínas auxiliares que regulam a atividade das chaperonas moleculares, como a Hsp90 e Hsp70, ajudando na maturação, estabilização e função de outras proteínas. No caso do GR, elas são essenciais para manter o receptor em um estado funcional antes da ligação do hormônio.
| |
Função das Cochaperonas no GR |
|
Manutenção do estado inativo do GR |
|
O GR fica no citoplasma ligado a um complexo de chaperonas e cochaperonas, principalmente Hsp90, Hsp70, Hop, p23 e FKBP51/FKBP52 |
|
Esse complexo impede a degradação do GR e mantém sua conformação pronta para se ligar ao glicocorticoide. |
|
Troca de cochaperonas na ativação do GR |
|
Quando o glicocorticoide se liga ao GR, ocorre uma troca de cochaperonas: FKBP51 é substituído por FKBP52, permitindo que o complexo se mova para o núcleo |
|
A proteína p23 estabiliza a interação do GR com a Hsp90, facilitando sua ativação |
|
Dissociação do complexo e ligação ao DNA |
|
No núcleo, o GR se dissocia das chaperonas e se liga ao DNA nos elementos responsivos a glicocorticoides (GREs), regulando a transcrição gênica |
O domínio C-terminal primariamente controla a atividade do receptor como um todo através de sua interação com as HSPs90 e ciclofilinas (quando o receptor está inativo) ou com hormônios e coativadores (quando o receptor está ativo). É o local onde ocorre a ligação da proteína de choque térmico que atua como molécula chaperona, que é a responsável, em parte, na manutenção da conformação inativa do GR, encobrindo os sinais de localização nuclear localizados nessa região.33
Como descrito, o receptor consiste de domínios distintos, incluindo o domínio N-terminal (DTN), o domínio de ligação de DNA central e o domínio de ligação do ligante C-terminal (LBD).
➥ Os corticoides por serem lipofílicos, atravessam facilmente a membrana celular por difusão passiva e ligam-se aos GRs que se encontram livres sem ligante no citosol como polímeros de baixo peso molecular (aproximadamente 90 kDa), parte de um complexo multiproteico inativo (aproximadamente 330 kDa), que consistem de duas moléculas HSP90, e outras proteínas como, p59, p60, p23, calreticulinas e imunofilinas da família FK 506
(FKBP51, FKBP52).34 As HSPs90 associadas ao GR parecem ser importantes na manutenção do GR (maturação) em uma conformação que seja apropriada para a ligação/ligante e para a inibição da translocação do GR para o núcleo (âncora citosólica). A ligação GR-HSP não permite a dimerização, a ativação e a ligação do complexo receptor-esteroide ao DNA. Após a ligação do ligante (hormônio), o GR é liberado desse complexo e translocado para o núcleo. Antes, entretanto, o complexo GR-chaperona se reorganiza novamente, e uma mudança conformacional do GR ocorre, determinando a exposição de duas sequências de localização nuclear (NLS).35 (Figura 5)
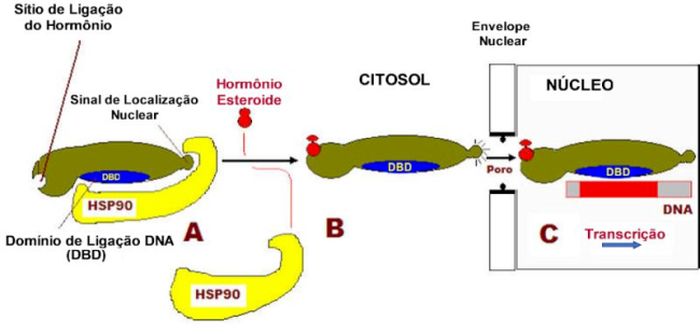
O transporte do GR para o núcleo da célula é mediado por duas NLSs – NLS1 e NLS2. A NLS2 está dentro do LBD e medeia uma transferência parcial do GR para o núcleo em vários tipos de células, enquanto que o NLS1 é um motivo básico curto no domínio dobradiça do GR (entre DBD (DNA binding domain) e LBD) que é suficiente para a completa transferência nuclear do receptor.36 Esta região é também o local de rotação das hélices do receptor, indispensável para permitir a alteração conformacional do complexo receptor-corticoide e é responsável pela especificidade do gene alvo, bem como pela alta afinidade de ligação ao DNA. Após o GR se liga a nucleoporinas e importinas que transportam o GR através do poro para o núcleo da célula.37,38
Como esperado, o GRα exerce sua atividade transcricional em genes responsivos a glicocorticoides por ligação a GREs localizados na região promotora desses genes como dímero, mas também como monômero ligando-se a outros fatores de transcrição.39 A sequência de ligação do GRE que induz a transcrição gênica é uma sequência palindômica de 15 pares de bases AGAACANNNTGTTCT (onde "n" é qualquer nucleotídeo) que estão ligados por homodímeros GR em um arranjo 'cabeça-a-cabeça'. Acredita-se que a ativação do gene alvo por GR ocorra principalmente por ligação direta a GREs e subsequente recrutamento de coativadores e histonas acetiltransferases.40
Para a repressão o GR se une às sequências de ligação de GR repetidas invertidas (IR-GBS) por tethering, por elementos compostos, competindo por sítios de ligação de DNA (BS), sequestrando fatores de transcrição (FTs) e competindo por cofatores com outros FTs.41 No entanto, descobertas recentes questionaram o modelo de transrepressão e sugerem que a ligação de GR ao DNA, não tethering, é necessária para a repressão gênica pró-inflamatória.42,43-45
A interação GR-DNA por meio de IR-GBS pode levar à inibição da expressão gênica através de uma variedade de mecanismos, incluindo competição com ativadores, recrutamento de correpressores, interferência com a maquinária de transcrição e modulação da cromatina.
Interações GR-DNA envolvem IR-GBS. A ligação a tal elemento leva à inibição da expressão gênica. Esses IR-nGREs têm uma sequência CTCC(N)0-2GGAGA de consenso e a análise estrutural mostrou que nesses locais dois monômeros GR se ligam nos lados opostos do DNA, em uma orientação cabeça-cauda e com cooperatividade negativa entre si.44,46
Além da ligação direta aos GREs, o complexo corticoide-GR pode interagir com outros fatores de transcrição, como AP-1 e NF-κB, modulando ainda mais a expressão gênica, reprimindo a transcrição de genes pró-inflamatórios. GR pode alterar o mRNA ou a estabilidade proteica de mediadores inflamatórios. (Figura 6)
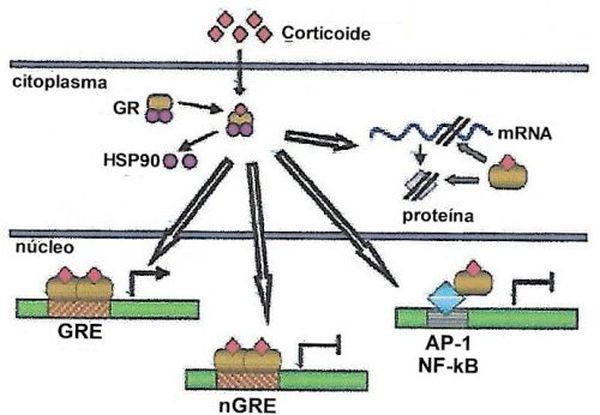
As mudanças na expressão gênica resultam em uma variedade de efeitos fisiológicos incluindo: redução da inflamação; supressão do sistema imunológico; regulação do metabolismo de carboidratos, proteínas e lipídios; efeitos sobre o sistema cardiovascular, sistema nervosos central e outros sistemas orgânicos. É importante que salientar que a resposta aos corticoides pode variar dependendo do tipo de célula, do tipo de tecido e da dose de medicamento.
Enquanto o papel de fatores gerais de transcrição na mediação da transcrição basal já está bem documentado, ficou estabelecido que os receptores nucleares recrutam fatores auxiliares chamados de correguladores: coativadores (proteínas nucleares que ativam a gene transcrição via atividade intrínseca histona acetiltransferase) e correpressores (proteínas nucleares que reprimem a gene transcrição e tem atividade histona desacetilase). Estes criam, dependendo do estado de ativação do receptor, uma ação permissiva (estimulação) ou repressora de transcrição junto ao promotor, e se comunicam com os fatores gerais de transcrição e com a RNA Polimerase II.9
As moléculas correguladoras do receptor de glicocorticoide (GR) são proteínas que modulam sua atividade transcricional após a ligação ao DNA. Elas podem ser classificadas em duas grandes categorias:
| |
Coativadores (Ativam a transcrição) |
| |
Facilitam a expressão gênica ao recrutar a maquinaria transcricional e modificar a cromatina. |
|
SRC-1, SRC-2 (TIF2/GRIP1) e SRC-3 (AIB1) ➢ Recrutam HATs, como CBP/p300, promovendo a acetilação das histonas e tornando a cromatina mais acessível. |
|
p300/CBP ➢ Cofatores essenciais que integram sinais e facilitam a ativação transcricional. |
|
CARM1 e PRMT1 ➢ Metiltransferases que modificam proteínas do complexo transcricional. |
| |
|
| |
Correpressores (Reprimem a transcrição) |
| |
Inibem a expressão gênica ao recrutar enzimas remodeladoras da cromatina. |
|
NCOR1 e NCOR2 (SMRT) ➢ Recrutam HDACs, fechando a cromatina e bloqueando a transcrição. |
|
GRIP1 (como correpressor em certos contextos) ➢ Pode atuar na repressão dependendo do gene e da interação proteica. |
✓ Se o GR ativa um gene, ele se liga a coativadores que promovem a abertura da cromatina.
✓ Se o GR reprime um gene, ele interage com correpressores que compactam a cromatina.
Vários grupos distintos de proteínas coativadoras têm sido identificados, com atuação na ativação transcricional mediada pelo GR.47 Estes coativadores constituem subunidades de grandes complexos multiproteicos que atuam em vários níveis funcionais, remodelando a cromatina, determinando modificação enzimática das histonas ou modulação do complexo de pré-iniciação via interação com a Pol II e GTFs (General Transcription Factors). A ligação dos receptores nucleares aos coativadores é frequentemente mediada por seus domínios de ativação transcricional. Os domínios AF-1 e AF-2 parecem ser os sítios de contato para o GR com coativadores, formando complexos proteicos envolvidos na ativação da transcrição.
A proteína de ligação CBP (cAMP response element binding protein (CREB) – binding protein) é um coativador nuclear ubíquo que funciona como integrador da gene transcrição, unindo ativadores de transcrição, tais como o GR, com o complexo basal de iniciação da transcrição. A CBP foi descoberta primeiramente como um coativador da CREB.48 Uma outra proteína relacionada, a chamada p300, apresenta homologia com a CBP. Ambas têm demonstrado formar interações proteína-proteína com membros do dispositivo basal transcricional incluindo a RNA polimerase II e fatores gerais de transcrição TFIIB (transcription factor II B) a proteína que se liga a TATA (TBP). Posteriormente ficou demonstrado ligarem-se a vários outros fatores de transcrição, como o AP-1 e NF-κB que upregulate vários genes inflamatórios da asma.
Quando os fatores de transcrição pró-inflamatórios NF-κB e AP-1 são ativados, eles se ligam a sequências específicas de reconhecimento no DNA e posteriormente também interagem com moléculas coativadoras como a p300/CBP e p300/CBP-associated factor (PCAF). Estas moléculas coativadoras atuam como um "interruptor" molecular que passa a controlar a gene transcrição. Todos têm atividade intrínseca de acetilação da histona, que resulta na acetilação dos núcleos da histona, reduzindo a sua carga.49
A identidade de correguladores que contribuem para a transativação de GR cresceu muito para números na casa das centenas.50 Alguns dos coativadores GR bem estudados são as proteínas da família SRC (steroid receptor coactivator), que consiste em três coativadores identificados: SRC-1, GRIP1 (glucocorticoid receptor-interacting protein 1),9,51,52 e p/CIP.53,54 Os GR após ativação pelos corticoides podem se ligar aos coativadores SRC que se ligam à CBP, resultando em aumento da transcrição. SRC e CBP apresentam atividade intrínseca HAT e são capazes de acetilar histonas.
Resultados de análise in vitro sugerem que os SRCs medeiam a atividade transcricional através de múltiplos mecanismos, incluindo:
1. A interação direta com receptores nucleares ligant-bound;9
2. Contato direto com determinados fatores gerais de transcrição, tais como TFIIB e TBP;55
3. Interação com os coativadores transcricionais comuns, tais como CBP, p300, e PCAF;9
4. Interação com outros coativadores tais como o CARM-1 (coactivator-associated arginine methyltransferase 1), ASC-2 (cancer-amplified transcriptional coactivator), PGC-1 (PPARg coactivator-1) e o SRA (steroid receptor RNA co-activator);56-58
5. Participação no remodelamento da cromatina através de sua atividade intrínseca da acetiltransferase da histona (HAT), que altera a conformação de nucleossomos na região promotor e permite a ligação de fatores necessários para aumentar a transcrição;59-61 e
6. Modificação enzimática de outros constituintes do complexo coativador.62
A ligação do GR ao dispositivo basal de transcrição não é a única função das proteínas coativadoras, que atuam na gene transcrição modulando a estrutura da cromatina. CDP-p300 e SRC-1 apresentam atividade HAT, alterando a conformação dos nucleossomos, que influencia o acesso ao promotor de fatores de transcrição, aumentando a transcrição.
Em contraposição, têm sido descritas proteínas com atividade HDAC, que reduzem a acetilação da histona e restauram a estrutura densa da cromatina, o que inibe a ligação de fatores de transcrição.
Receptores ativados de glicocorticoides podem se ligar diretamente à CBP ou outros coativadores para inibir a atividade HAT, revertendo o desenrolar do ADN ao redor do núcleo da histona, remodelando a cromatina e assim reprimir os genes inflamatórios. Baixas concentrações de corticoides ativam os receptores de corticoides, recrutam HDACs (principalmente HDAC1 e 2) para ativar o complexo transcricional, resultando na desacetilação das histonas, reduzindo a transcrição de genes inflamatórios e aumentando a transcrição de genes anti-inflamatórios.49
Um conceito sugere que os ativadores da transcrição, como o GR, devam agir também em uma outra enzima que atue no remodelamento da cromatina, além dos coativadores com atividade HAT. Estes complexos remodeladores cromatina ATP-dependentes são conhecidos como Swi/Snf (switch/sucrose nonfermentable). O complexo Swi/Snf hidrolisa o ATP e utiliza a energia da hidrólise do ATP para romper interações histona-DNA e remodelar a cromatina, permitindo que os fatores da transcrição se liguem ao promotor.63 Embora o exato mecanismo pelo qual o Swi/Snf altera a estrutura da cromatina não esteja de todo elucidado, já existem evidências de que o recrutamento do Swi/Snf é essencial para a regulação da transcrição do gene, incluindo aqueles mediados pelo GR.64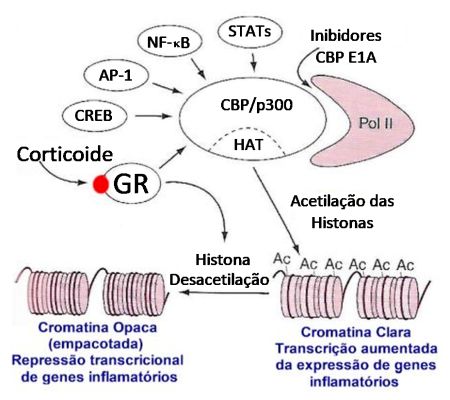
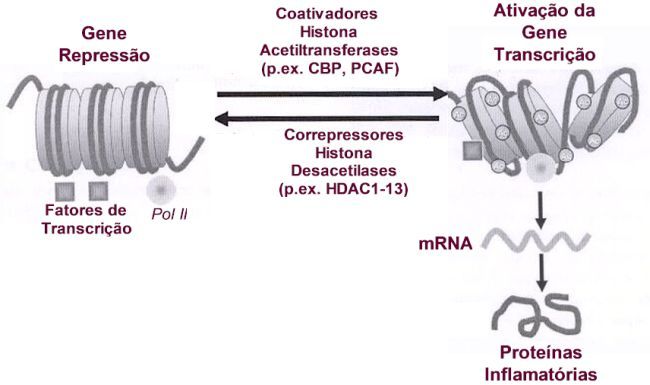
A Figura 7 sintetiza o que foi exposto. Nela são evidenciados os múltiplos fatores de transcrição que se ligam às moléculas coativadoras, como a CBP ou p300, as quais apresentam atividade intrínseca histona acetiltransferase, resultando na acetilação das histonas, ao redor das quais o DNA está enrolado. A acetilação determina remodelamento da cromatina com a sua abertura, permitindo o acesso aos fatores de transcrição, dentre eles o GR, resultando no aumento da gene transcrição, produzindo mRNAs responsáveis pela síntese de proteínas.
Por outro lado, os receptores de glicocorticoides após a ativação pelos corticoides interagem na gene transcrição por outras duas formas: 1) podem se ligar a coativadores dos receptores de glicocorticoide SRC que se ligam à CBP resultando em aumento da transcrição ou; 2) se ligam diretamente à CBP, resultando desacetilação das histonas, compactando o DNA, impedindo desta forma a ligação de fatores de transcrição, determinando redução na expressão gênica.
| Uma questão importante: os corticoides desativam apenas os genes inflamatórios. Eles nitidamente não suprimem a atividade de todos os genes e são bem tolerados como terapêutica. |
|
Os receptores de corticoides provavelmente se ligam unicamente aos coativadores que são ativados por fatores de transcrição pró-inflamatórios, como o NF-κB e AP-1, embora não se saiba como ocorre esta ação específica de reconhecimento. A exploração da especificidade tecidual da ligação do GR surgiu como uma estratégia terapêutica potencial para reter efeitos benéficos específicos do tratamento com corticoide, como a imunomodulação, ao mesmo tempo em que previne efeitos adversos, como obesidade, diabetes e osteoporose.65
A maioria dos efeitos anti-inflamatórios dos glicocorticoides parece resultar de um importante mecanismo regulador negativo chamado transrepressão, no qual o GR ligado ao ligante é recrutado para a cromatina por interações proteína-proteína com fatores de transcrição ligados ao DNA, particularmente NF-κB e AP-1. Tanto o NF-κB quanto o AP-1 são fatores de transcrição que amplificam, coordenam e perpetuam mecanismos de ativação de múltiplos genes inflamatórios.66
➭O complexo corticoide-GR pode controlar a transcrição de genes por vários mecanismos, dentre eles:
■ Por controle direto ou indireto sobre a transcrição de certos genes-alvo.67,68 O número de genes controlados diretamente pelo corticoide por célula é estimado entre 10 e 100, sendo que outros são regulados indiretamente através de interação com diferentes fatores de transcrição.1 A maioria dos efeitos anti-inflamatórios dos corticoides resulta de um importante mecanismo regulador negativo chamado transrepressão, no qual o GR ligado ao ligante é recrutado para a cromatina por interações proteína-proteína com fatores de transcrição ligados ao DNA, particularmente NF-κB e proteína ativadora-1 (AP-1).
■ Por interação direta proteína-proteína com o fator de transcrição AP-1 e o GR.69 Funcionalmente, as consequências destas interações resultam em uma mútua repressão transcricional tanto do AP-1 como do GR70,71 o que inibe os efeitos pró-inflamatórios de uma série de citocinas.
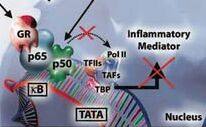 ■ Por repressão direta da transcrição dos fatores nucleares pró-inflamatórios NF-κB72,73 e NF-AT (nuclear factor of activated T-cells) que regulam a expressão de outros genes alvo inflamatórios. O GR interage com o NF-κB pela ligação à subunidade p65 do heterodímero NF-κB,74 impedindo a ligação DNA NF-κB,75,76 suprimindo a transcrição de genes como os das citocinas IL-6 e IL-8,75,77 de enzimas inflamatórias e moléculas de adesão (Figura 8 ).
■ Por repressão direta da transcrição dos fatores nucleares pró-inflamatórios NF-κB72,73 e NF-AT (nuclear factor of activated T-cells) que regulam a expressão de outros genes alvo inflamatórios. O GR interage com o NF-κB pela ligação à subunidade p65 do heterodímero NF-κB,74 impedindo a ligação DNA NF-κB,75,76 suprimindo a transcrição de genes como os das citocinas IL-6 e IL-8,75,77 de enzimas inflamatórias e moléculas de adesão (Figura 8 ).
■ Por aumento das ribonucleases das células, e desta forma reduzindo os níveis de mRNA.78
01.Barnes PJ. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br J Pharmacol 2006; 148:245-54.
02.Barnes PJ, Adcok IM. Transcription factors and asthma. Eur Respir J 1998; 12:221.
03.Farah, SB.– DNA Segredos & Mistérios. São Paulo: Sarvier; 2000.
04.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. et al. — Biologia Molecular da Célula. 6 ª edição. Porto Alegre: Artmed; 2017.
05.Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell 1998; 92:307-13.
06.Struhl K, Moqtaderi Z. The TAFs in the HAT. Cell 1998; 94:1-4.
07.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone aceyltransferases. Cell 1996; 87:953-9.
08.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem 2001; 70:81-120.
09.McKenna NJ, Rainer BL, O'Malley BE. Nuclear receptor coregulators: cellular and molecular biology. Endocr.Rev 1999; 20:321-44.
10.Hollenberg SM, Weinberger C, Ong ES, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985; 318:635641.
11.Evans RM. The steroid and thyroid hormone receptor superfamily. Science 1988; 247:889-895.
12.Beato M, Truss M, Chavez S. Control of transcription by steroid hormones. Ann N Y Acad Sci 1996; 784:93-123.
13.Beato M, Herrrkich P, Schütz G, Steroid hormone receptors: many actors in search of a plot. Cell 1995; 83:851-857.
14.Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J 2002; 16:61-71.
15.Nicolaides NC, Chrousos G, Kino T. Glucocorticóide Receptor. [Atualizado em 21 de novembro de 2020]. In: Feingold KR, Anawalt B, Blackman MR, et al., edi. Endotexto [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK279171/
16.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985; 318:635-641.
17.Lu NZ, Cidlowski JA. The origin and functions of multiple human glucocorticoid receptor isoforms. Ann N Y Acad Sci 2004; 1024:102-123.
18.Vandevyver S, Dejager L, Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr Rev 2014; 35:671-93.
19.Dieken ES, Miesfeld RL. Transcriptional transactivation functions localized to the glucocorticoid receptor N terminus are necessary for steroid induction of lymphocyte apoptosis. Mol Cell Biol 1992; 12:589597.
20.O’Malley B. The steroid receptor superfamily: more excitement predicted for the future. Mol Endocrinol 1990; 4:363–369.
21.Danielian PS, White R, Lees JA, Parker MG. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J 1992; 11:1025–1033.
22.Kumar R, Thompson EB. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol Endocrinol 2003; 17:1–10.
23.Hollenberg SM, Evans RM. Multiple and cooperative trans-activation domains of human glucocorticoid receptor. Cell 1988; 55:899-906.
24.Ford J, McEwan IJ, Wright AP, Gustafsson JA. Involvement of the transcription factor IID protein complex in gene activation by N-terminal transactivation domain of glucocorticoid receptor in vivo. Mol Endocrinol 1997; 11:1467-75.
25.Härd T, Kellenbach E, Boelens R, et al. Solution structure of the glucocorticoid receptor DNA-binding domain. Science 1990; 249:157–160.
26.Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991; 352:497–505.
27.Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol 2013; 20:876–883.
28.Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol 2005; 94:383-94.
29.Beck IM, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Ver 2009; 30:830-82.
30.Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr Opin Cell Biol 1997; 9:222-32.
31.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002; 110:93-105.
32.Liberman AC, Budziñski ML, Sokn C, Gobbini RP, Steininger A, Arzt E. Regulatory and Mechanistic Actions of Glucocorticoids on T and Inflammatory Cells. Front Endocrinol (Lausanne). 2018; 9:235. doi: 10.3389/fendo.2018.00235.
33.Loke TK, Corrigan CJ, Lee TH. Glucocorticoid Effects on Mediator Modulation. In : Jan M. Agosti and Albert L. Sheffer. Biotherapeutic Approaches to Asthma . New York: Marcel Dekker; 2002:327-352.
34.Cheung J, Smith DF. Molecular chaperone interactions with steroid receptors: an update. Mol Endocrinol 2000l; 14:939-46.
35.Riggs DL, Roberts PJ, Chirillo SC, Cheung-Flynn J, Prapapanich V, Ratajczak T, Gaber R, Picard D, Smith DF. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J 2003; 22:1158-67.
36.Tang Y, Getzenberg RH, Vietmeier BN, Stallcup MR, Eggert M, Renkawitz R, DeFranco DB. The DNA-binding and tau2 transactivation domains of the rat glucocorticoid receptor constitute a nuclear matrix-targeting signal. Mol Endocrinol 1998; 12:1420-31.
37.Freedman ND, Yamamoto KR. Importin 7 and importin alpha/importin beta are nuclear import receptors for the glucocorticoid receptor. Mol Biol Cell 2004; 15:2276-86.
38.Echeverría PC, Mazaira G, Erlejman A, Gomez-Sanchez C, Piwien Pilipuk G, Galigniana MD. Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol Cell Biol 2009; 29:4788-97.
39.Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev 1996; 17:245-61.
40.Muzikar KA, Nickols NG, Dervan PB. Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc Natl Acad Sci U S A 2009; 106:16598-603.
41.Timmermans S, Souffriau J, Libert C. A General Introduction to Glucocorticoid Biology. Front Immunol 2019;10:1545.
doi: 10.3389/fimmu.2019.01545.
42.Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, Schwalie P, Hübner N, Evans RM. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell 2013; 49:158-71.
43.Weikum ER, de Vera IMS, Nwachukwu JC, Hudson WH, Nettles KW, Kojetin DJ, Ortlund EA. Tethering not required: the glucocorticoid receptor binds directly to activator protein-1 recognition motifs to repress inflammatory genes. Nucleic Acids Res 2017; 45:8596-8608.
44.Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 2011; 145:224-41.
45.Gebhardt JC, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, Maniatis T, Xie XS. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods 2013; 10:421-6.
46.Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol 2013; 20:53-8.
47.Featherstone M. Coactivators in transcription initiation: here are your orders. Curr Opin Genet Dev 2002r; 12:149-55.
48.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 2001; 114:2363-73.
49.Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol. 2011; 163:29-43.
50.Ramamoorthy S, Cidlowski JA. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum Dis Clin North Am 2016; 42:15-31,vii. doi: 10.1016/j.rdc.2015.08.002.
51.Hong H, Kohli K, Trivedi A, Johnson DL, Stallcup MR. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci U S A 1996; 93:4948-52.
52.Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J 1996; 15:3667-75.
53.Takeshita A, Cardona GR, Koibuchi N, Suen CS, Chin WW. TRAM-1, A novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid receptor coactivator-1. J Biol Chem 1997; 272:27629-34.
54.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 1997; 387:677-84.
55.Takeshita A, Yen PM, Misiti S, Cardona GR, Liu Y, Chin WW. Molecular cloning and properties of a full-length putative thyroid hormone receptor coactivator. Endocrinology 1996; 137:3594-7.
56.Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science 1999; 284:2174-7.
57.Lee SK, Anzick SL, Choi JE, Bubendorf L, Guan XY, Jung YK, Kallioniemi OP, Kononen J, Trent JM, Azorsa D, Jhun BH, Cheong JH, Lee YC, Meltzer PS, Lee JW. A nuclear factor, ASC-2, as a cancer-amplified transcriptional coactivator essential for ligand-dependent transactivation by nuclear receptors in vivo. J Biol Chem 1999; 274:34283-93.
58.Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, O'Malley BW. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999; 97:17-27.
59.Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell 1998; 92:307-13.
60.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 1997; 90:569-80.
61.Spencer TE; Jenster G; Burcin MM; Allis CD; Zhou J; Mizzen CA; McKenna NJ; Onate SA; Tsai SY; Tsai MJ; O'Malley BW. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature 1997; 389:194-8.
62.Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 1999; 98:675-86.
63.Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 2000; 20:1899-910.
64.Fryer CJ, Archer TK. Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature 1998; 393:88-91.
65.Syed AP, Greulich F, Ansari SA, Uhlenhaut NH. Anti-inflammatory glucocorticoid action: genomic insights and emerging concepts. Curr Opin Pharmacol 2020; 53:35-44.
66.Adcock IM, Caramori G. – Transcription Factors. In: Peter J. Barnes. Asthma and COPD. Amsteram: Elsevier; 2009:373--380.
67.Gronemeyer H. Control of transcription activation by steroid hormone receptors. FASEB J 1992; 6:2524-9.
68.Barnes JB, Rodger IW, Thomson NC. Asthma - Basic Mechanisms and Clinical Management . London, Academic Press, 3rd ed., 1998.
69.Ponta H, Cato ACB, Herrlick P. Interference of specific transcription factors. Biochim Biophys Acta 1992; 1129:255-61.
70.Horvath G, Wanner A. Tracheobronchial Circulation. In: Barnes P, Drazen J, Rennard S, Thomson N. Asthma and COPD Basic Mechanisms and Clinical Management. London: Academic Press; 2002:177-182.
71.Nauck M, Roth M, Tamm M et al. Induction of vascular endothelial growth factor by platelet-activating factor and platelet-derived growth factor is downregulated by corticosteroids. Am J Respir Cell Mol Biol 1997; 16:398-406.
72.Adock IM, Brown CR, Gelder CM, Shirasaki H, Peters MJ, Barnes PJ. The effects of glucocorticoids on transcription factor activation in human peripheral blood mononuclear cells. Am J Physiol 1995; 37:C331-8.
73.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF- 6 B and the glucocorticoid receptor. Proc Natl Sci USA 1994; 91:752-6.
74.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell 1995; 83:851-7.
75.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF 6 B and the glucocorticoid receptor. Proc Natl Acad Sci USA 1994; 91:752-6.
76.Adock IM, Shirasaki H, Gelder CM, et al. The effects of glucocorticoids on phorbol ester and cytokine stimulated transcription factor activation in human lung. Life Sci 1994; 55:1147-53.
77.Mukaida N, Morita M, Ishikawa Y et al. Novel mechanism of glucocorticoid-mediated gene repression: nuclear factor- 6 B is target for glucocorticoid-mediated interleukin 8 gene repression. J Biol Chem 1994; 269:13289-95.
78.O'Byrne P. Corticosteroids. In: Albert RK, Spiro SG, Jett JR. Comprehensive Respiratory Medicine .London:Mosby,1999.
79.Ristimaki A, Narko K, Hla T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J 1996; 318:325-31.
80.Bickel M, Iwai Y, Pluznik DH, Cohen RB. Binding of sequence-specific proteins to the adenosine- plus uridine-rich sequences of the murine granulocyte/macrophage colony-stimulating factor mRNA. Proc Natl Acad Sci USA 1992; 89:10001-5.
81.Adkins KK, Levan TD, Miesfeld RL, Bloom JW. Glucocorticoid regulation of GM-CSF; evidence for transcriptional mechanisms in airway epithelial cells. Am J Physiol 1998; 275: L372-8.
82.
Newton R, Seybold J, Kuitert LM, Bergmann M, Barnes PJ. Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J Biol Chem 1998; 273:32312-21.
83.Wang J, Zhu Z, Nolfo R, Elias JA. Dexamethasone regulation of lung epithelial cell and fibroblast interleukin-11 production. Am J Physiol 1999; 276:L175.
84.Bergmann M, Barnes PJ, Newton R. Molecular regulation of granulocyte macrophage colony-stimulating factor in human lung epithelial cells by interleukin (IL)-1B, IL-4, and IL-13 involves both transcriptional and post-transcriptional mechanisms. Am J Cell Mol Biol 2000; 22:582-9.
85.Caelles C, Gonzalez-Sancho JM, Monoz A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev 1997; 11:3351-64.
86.Vanden Berghe W, Vermeulen L, De Wilde G, De Bosscher K, Boone E, Haegeman G. Signa trasnduction by tumor necrosis factor and gene regulation of the inflammatory cytokine interleukin-6. Biochem Pharmacol 2000; 60:1185-95.
87.Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol 2001; 21:771-80.
88.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein-kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol 2002; 22:7802-11.
89.Lewis GD, Campbell WB, Johanson AR. Inhibition of prostaglandin synthesis by glucocorticoids in human endothelial cells. Endocrinology 1986; 119:62-9.
90.Chung KF, Wiggins J, Collins J. Corticosteroids. In: Bronchial asthma: mechanisms and therapeutics. Boston, Little Brown and Company, 3rd ed., 1993.
91.Abbinante-Nissen JM, Simpson LG, Leikauf GD. Corticosteroids increases secretory leukocyte protease inhibitor transcript levels in airway epithelial cells. Am J Physiol 1995; 12:L601-6.
92.Borson DB, Gruenert DC. Glucocorticoids induce neutral endopeptidase in transformed human tracheal epithelial cells. Am J Physiol 1991; 260:L83-9.
93.Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol 2011; 163:29-43.
94.Wang P, Wu P, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor kappa B activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem 1995; 270:9558-63.
95.Chernoff AE, Granowitz EV, Shapiro L et al. A randomized, controlled trial of IL-10 in humans. Inhibition of inflammatory cytokine production and immune responses. J Immunol 1995; 154:5492-9.
96.Naseer T, Minshall EM, Leung DY, et al. Expression of IL-12 and IL-13 mRNA in asthma and their modulation in response to steroid therapy. Am J Respir Crit Care Med 199; 155:845-51.
97.Levine SJ, Benfield T, Shelhamer JH. Corticosteroids induce intracellular interleukin-1 receptor antagonist type I expression by a human airway epithelial cell line. Am J Respir Cell Mol Biol 1996; 15: 245-51.
98.Sousa AR, Lane SJ, Nakhosteen JA, Lee TH, Poston RN. Expression of interleukin-1 beta (IL-1 b ) and interleukin-1 receptor antagonist (IL-1ra) on asthmatic bronchial epithelium. Am J Respir Crit Care Med 1996; 154: 1061-6.
99.Colotta F, Re F, Muzio M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science (Washington DC) 1993; 261:4725.
100.Mak JCW, Nishikawa M, Barnes PJ. Glucocorticosteroids increase $ 2 -adrenergic receptor transcription in human lung. Am J Physiol 1995; 12:L41-6.
101.Adocck IM. Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther 2001; 14:211-9.
102.Ito K, Barnes PJ, Adock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits IL-1B-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20:6891-903.
103.Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem 2001; 276:29603-10.
104.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phsophatase-mediated inhibition of MAPK p38. Mol Cell Biol 2002; 22:7802-11.
105.Gillis S, Crabtree GR, Smith KA. Glucocorticoids-induced inhibition of T-cell growth factor production: I. The effect on mitogen-induced lymphocyte proliferation. J Immunol 1979; 123:1624-31.
106.Nickel R, Beck LA, Stellato C, Schleimer RP. Chemokines and allergic disease. J Allergy Clin Immunol 1999; 104:723-42.
107.Lamas AM, Leon OG, Schleimer RP. Glucocorticoids inhibit eosinophil responses to granulocyte macrophage colony-stimulating factor. J Immunol 1994; 147:254-9.
108.Wallen IL, et al. Glucocorticoids inhibit cytokine-mediated eosinophil survival. J Immunol 1991; 147:3490-5.
109.Altman LC, et al. Effect of corticosteroids on eosinophil chemotaxis and adherence. J Clin Invest 1981; 67:28-36.
110.Colotta F, Re F, Muzio M, et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulate by IL-4. Science 1993; 261:472-5.
111.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leucocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA 1992; 89:9991-5.
112.Weiss EB, Stein M. Bronchial Asthma. Little Brown and Company, 3rd ed., Boston, 1993.
113.Adock IM, Peters M, Gelder C, Shirasaki H, Brown CR, Barnes PJ. Increased tachykinin receptor gene expression in asthmatic lung and its modulation by steroids. J Mol Endocrinol 1993; 11:1-7.
114.Knowles RG, Salter M, Brooks SL, Moncada S. Anti-inflammatory glucocorticosteroids inhibit the induction by endotoxin of nitric oxide synthase in the lung, liver and aorta of the rat. Biochem Biophys Res Commun 1990; 172:1042-8.
115.Nelson BV, Sears S, Woods J, Ling CY, Hunt J, Clapper LM, Gaston B. Expired nitric oxide as a marker for childhood asthma. J Pediatr 1997; 130:423-7.
116.Baraldi E, Azzolin NM, Zanconato S, Dario C, Zacchello F. Corticosteroids decrease exhaled nitric oxide in children with acute asthma. J Pediatr 1997; 131:381-5.
117.Mitchell JA, Belvisi MG, Akarasereenont P, Robbins RA, Kwon OJ, Croxtall J, Barnes PJ, Vane JR. Induction of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. Br J Pharmacol 1994; 113:1008-14.
118.Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kB and nuclear factor-interleukin 6 in the tumor necrosis- a -dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem 1995; 270: 31315-20.
119.Newton R, Kuitert LM, Slater DM, Adcock IM, Barnes PJ. Cytokine induction of cytosolic phospholipase A2 and cyclooxygenase-2 mRNA is suppressed by glucocorticoids in human epithelial cells. Life Sci. 1997; 60:67-78.
120.Yanagisawa M, Kurihara H, Kimura, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332:411-5.
121.Vittori E, Marini M, Fasoli A. Increased expression of endothelin in bronchial epithelial cells of asthmatic patients and effect of corticosteroids. Am Rev Respir Dis 1992; 146:1320-5.
122.Reisman D, Thompson EA. Glucocorticoid regulation of cyclin D3 gene transcription and mRNA stability in lymphoid cells. Mol Endocrinol 1995; 9:1500-9.